


Mariana Morais Tavares Colferai; Elizabeth Leocadia Fernandes; Denise Steiner; Gabriela Momente Miquelin; Camila Carneiro Marques; Kelly Cristina Signor
O sarcoma de Kaposi é neoplasia multicêntrica rara originária de células endoteliais com manifestação cutânea e extracutânea. Descreve-se o caso de variante clínica queloidiana de SK, incomum, em paciente do sexo masculino, de 32 anos, portador da síndrome da imunodeficiência adquirida (Aids), com regressão ao tratamento combinado de terapia antirretroviral e radioterapia.
Keywords: sarcoma de Kaposi; herpes-vírus humano 8; síndrome de imunodeficiência adquirida; queloide
Descrito em 1872, o sarcoma de Kaposi (SK) é neoplasia multicêntrica rara originária de células endoteliais com manifestação cutânea e extracutânea.1
Relata-se o caso de paciente portador de rara variante de SK epidêmico, a queloidiana, com regressão ao tratamento combinado de terapia antirretroviral e radioterapia.
Paciente do sexo masculino, de 32 anos, branco, solteiro, empresário, procurou atendimento médico referindo surgimento de lesão assintomática no membro inferior esquerdo há cerca de seis meses, com crescimento progressivo. Negava trauma local, assim como o uso de medicações sistêmicas e tópicas. Apresentava antecedente de hérnia discal e alergia ao ácido acetilsalicílico, dipirona e anti-inflamatório não esteroidal.
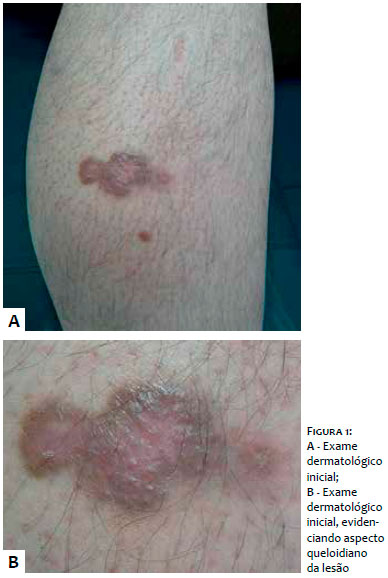
Ao exame dermatológico, apresentava nódulo eritêmato-violáceo, endurecido, indolor à palpação, localizado na borda interna da perna esquerda. (Figura 1)
Nos exames laboratoriais, constatou-se HIV positivo, com carga viral inicial de 378.000, CD4: 324 e CD8: 565.
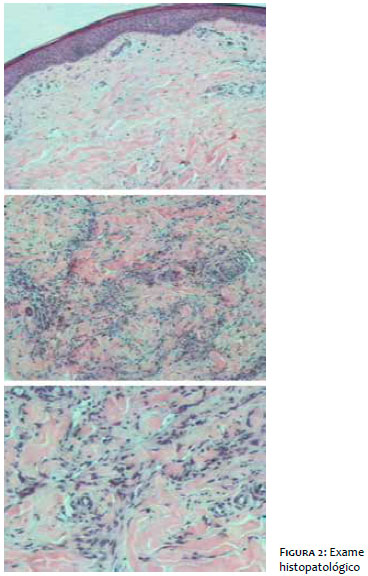
Ao exame histopatológico da lesão (Figura 2), a epiderme exibia retificação de cristas epiteliais, derme superficial e profunda com numerosos vasos pequenos com paredes colabadas e endotélio tumefeito, em meio a células fusiformes com nucléolos evidentes. Observavam-se focos de hemácias extravasadas, focos de deposição de pigmento hemossiderótico, múltiplos focos de infiltrado misto e espessamento intersticial por fibrose. O diagnóstico foi compatível com SK.
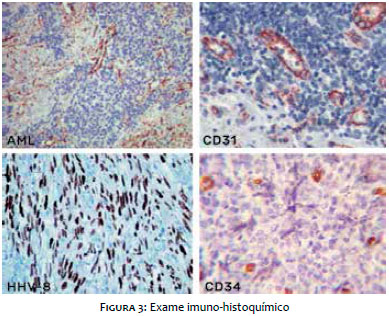
O exame imuno-histoquímico (Figura 3) mostrou positividade para actina de músculo liso (AML), CD31, CD34 e herpes-vírus 8.
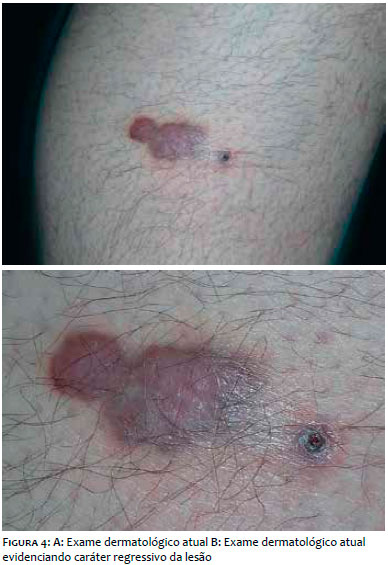
Iniciou-se o acompanhamento multidisciplinar com as clínicas de oncologia e infectologia, assim como a terapia antirretroviral. Após três meses de tratamento, cursava com carga viral: 91, CD4: 503 e CD8: 933, porém com regressão parcial da lesão. Foi então submetido a dez sessões de radioterapia, apresentando melhora importante. (Figura 4)
Após dois anos de acompanhamento, o novo exame histopatológico da lesão revelou ausência de critérios histológicos para o diagnóstico de SK.
Moritz Kaposi descreveu, na Hungria, cinco homens com um tipo de sarcoma multifocal incomum da pele, que ele denominou “sarcoma pigmentado idiopático múltiplo da pele”. Essa entidade patológica ficou conhecida como sarcoma de Kaposi(SK).2
O SK é tumor endotelial multicêntrico, de baixo grau de malignidade, considerado raro até o início da epidemia de Aids. A partir de 1981, sua incidência aumentou consideravelmente, sobretudo em pacientes homens que fazem sexo com homens (SK epidêmico associado ao HIV). Em 1994, descobriu-se a participação de um vírus do grupo herpes (HHV-8 ou SKHV) na etiopatogenia do SK. Estudos mais recentes mostram três fatores mais implicados em sua etiopatogenia: a infecção pelo HIV, a infecção pelo HHV-8 e o papel das citocinas. A partir de 1996, nos pacientes com infecção pelo HIV, a introdução da terapia antirretroviral combinada (Haart) resultou em redução significativa do número de novos casos de SK epidêmico.
Desde sua descrição, a doença foi relatada em cinco quadros clínicos diferentes com apresentação, epidemiologia e prognóstico distintos. Os cinco subtipos são: 1) SK clássico, doença indolente vista principalmente em homens de meia-idade do sul ou leste europeu; 2) SK cutâneo endêmico africano , um processo localizado agressivo afetando indivíduos de meia-idade na África tropical; 3) SK linfadenopático endêmico africano, doença agressiva que afeta pacientes jovens, especialmente crianças com menos de dez anos de idade; 4) SK em pacientes imunossuprimidos pela Aids (epidêmico); ou 5) secundário ao linfoma ou ao tratamento imunossupressor.3
Pode limitar-se à pele, mas pode acometer mucosa oral, linfonodos e vísceras. Pode ter evolução indolente, apenas com lesões cutâneas restritas aos membros inferiores, ou progressão rápida, com lesões cutâneas extensas e viscerais.
O SK associado à Aids inicia-se com múltiplos nódulos na região superior do tórax, cabeça e pescoço e evolui rapidamente com disseminação na pele e para os órgãos internos, levando à morte.
As formas clínicas evolutivas de apresentação do SK são: (1) SK macular, (2) SK em placa localizada não destrutiva, (3) SK exofítico, (4) SK infiltrativo, (5) SK linfadenopático generalizado, (6) SK disseminado cutâneo e visceral, (7) SK telangiectásico, (8) SK queloidiano, (9) SK equimótico e (10) SK cavernoso ou linfangioma-like. O SK extracutâneo é mais evidente no trato gastrointestinal, linfonodos e pulmões.4
A histopatologia varia de acordo com o desenvolvimento das lesões.
1) Lesões maculares: há proliferação, na derme superficial, de espaços vasculares revestidos por células endoteliais que separam os feixes colágenos e se acompanham de infiltrado discreto de linfócitos e plasmócitos.
2) Lesões em placas: as alterações vasculares estendem-se para a derme profunda e subcutâneo, e surgem células fusiformes que são positivas para marcadores histoquímicos de vasos.
3) Lesões nodulares: predominam as células fusiformes com atipias nucleares e mitose, formando feixes, e, na periferia, observam-se espaços vasculares bizarros contendo eritrócitos, que se apresentam também extravasados, e há macrófagos contendo hemossiderina em meio a infiltrado de linfócitos, plasmócitos, histiócitos e, esporadicamente, neutrófilos.
A descrição da variante queloidiana é extremamente incomum e limitada a um relato de 1994 de três casos. As lesões são firmes e com consistência de borracha, e podem ser lineares. Histologicamente, há notável expansão dérmica com colágeno denso e hialinizado com apresentação distinta de queloides. O diagnóstico diferencial histológico inclui cicatriz no local de biópsia cutânea anterior a alguma lesão de SK. É postulado que as citocinas desempenham papel fundamental na evolução das mudanças do estroma queloidiano nessa variante incomum.5
A diagnose é clínica e histopatológica. Na diagnose diferencial, devem ser considerados angiomas e outros tumores vasculares. As formas disseminadas da Aids exigem diferenciação com angiomatose bacilar, angiomas, metástases, líquen plano, sífilis, nevos melanocíticos e picadas de inseto.6
O SK apresenta boa resposta às diversas estratégias terapêuticas. Para lesões localizadas, excisão cirúrgica, crioterapia e radioterapia podem ser empregadas. Para lesões cutâneas maiores, múltiplas ou acometendo vísceras, o tratamento sistêmico está indicado. Entre eles, nos casos de doença disseminada, a quimioterapia é utilizada para atuar tanto nas lesões cutâneas como nas viscerais. Existem diversos agentes quimioterápicos ativos, com taxa de resposta variável de 60 a 80%, entre eles: as antraciclinas lipossomais (doxorrubicina, daunorrubicina) o paclitaxel a vimblastina e o etoposide. A imunoterapia com interferon pode ser indicada em casos selecionados.1 A introdução da terapêutica antirretroviral altamente ativa, permitindo a reconstituição imunológica dos doentes infectados pelo HIV, refletiu profundamente no SK ligado a essa infecção, diminuindo, de modo significativo, sua ocorrência; ao ser introduzida em doentes com HIV e SK, também possibilita a regressão do tumor.6
Mariana Morais Tavares Colferai:
Revisão bibliográfica, formulação do artigo, submissão do artigo
Gabriela Momente Miquelin:
Revisão bibliografica, formulaçao do artigo
Elizabeth Leocadia Fernandes:
Orientaçao, discussão e correçao do artigo
Denise Steiner:
Orientação e revisão final
Kelly Cristina Signor:
Registro fotográfico e correção das fotografias
Camila Carneiro Marques:
Revisão bibliografica e correção ortografica
1. Ohe EMDN, Padilha MHVQ, Enokihara MMSS, Almeida FA, Porro AM. Fatal outcome in classic Kaposi's sarcoma. An Bras Dermatol. 2010;85(3):375-9.
2. Bolognia JL, Jorizzo JL, Rapini RP . Dermatology. 2nd ed. Rio de Janeiro: Elsevier; 2011.
3. James WD, Berger TG, Elston DM. Andrews' diseases of the skin: clinical dermatology. 10th ed. Rio de Janeiro: Elsevier; 2007.
4. Belda Junior W, Di’Chiacchio N, Criado PR. Tratado de Dermatologia. Vol 1. São Paulo: Editora Atheneu; 2010.
5. Grayson W, Pantanowitz, L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008; 3:31
6. Sampaio SAP, Rivitti EA. Dermatology. 3rd ed. rev. e ampl. São Paulo: Artes médicas; 2007.
Trabalho realizado na Universidade de Mogi das Cruzes – Mogi das Cruzes (SP), Brasil.
 All content the journal, except where identified, is under a Creative Commons Attribution-NonCommercial 4.0 International license - ISSN-e 1984-8773
All content the journal, except where identified, is under a Creative Commons Attribution-NonCommercial 4.0 International license - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}