


Anna Carolina Brandão Vasconcelos; John Verrinder Veasey
Data de recebimento: 29/08/2019
Data de aprovação: 18/08/2020
Trabalho realizado na Clínica de Dermatologia do Hospital da Santa Casa de São Paulo, São Paulo (SP), Brasil.
Suporte Financeiro: Nenhum.
Conflito de Interesses: Nenhum.
A doença de Dowling-Degos (DDD) é uma genodermatose rara, caracterizada principalmente por hiperpigmentação reticular progressiva de áreas flexurais, que pode estar associada a um grande espectro de lesões benignas e neoplasias cutâneas. Pode ocasionar prejuízo psicossocial devido ao potencial de deformidade das lesões e dano estético significativo. Apresentamos dois casos de pacientes, ambos do sexo masculino, com máculas hipercrômicas, múltiplos comedões, cistos epidérmicos, cicatrizes cribriformes em: face, região cervical, dorso, tórax anterior, axilas e região genital, além de tumorações desfigurantes, características da DDD.
Keywords: Carcinoma; Dermatopatias; Genética; Hidradenite; Hidradenite Supurativa; Pigmentação da Pele; Síndrome
A doença de Dowling-Degos (DDD) é uma genodermatose rara, de transmissão autossômica dominante, com penetrância e expressividade variáveis, descrita inicialmente por Jones e Grice em 1974.1,2
Trata-se de doença de surgimento tardio, após a segunda década de vida, caracterizada por discromia reticulada, principalmente em face e superfícies flexurais como pescoço, axilas, fossas antecubitais, áreas submamárias e virilhas.3,4 Concomitantemente, podem ocorrer lesões da unidade pilossebácea como comedões, cistos epidérmicos, abscessos, hidradenite supurativa, além de neoplasias cutâneas, como carcinoma espinocelular e ceratoacantoma.2,4 A pigmentação é progressiva e simétrica, geralmente extensa e assintomática, exacerbada pela exposição solar.5,6,7
Diagnósticos diferenciais devem ser feitos com acantose nigricante, acropigmentação de Kitamura, doença de Galli-Galli, discromatose universal hereditária e discromatose simétrica hereditária.8 Tais doenças podem apresentar sobreposições clínicas entre si, sendo que alguns autores as consideram enfermidades separadas, enquanto outros, espectro de uma mesma doença.4,5,9
O diagnóstico é baseado nas características clínicas sugestivas, associadas aos achados no exame histopatológico.7,8 A histopatologia mostra hiperpigmentação da camada basal, proliferações filiformes da epiderme, por vezes semelhantes a chifre de rena, além de hiperqueratose e brotamentos, surgindo a partir do infundíbulo pilar, caracterizando um “plug” folicular.1,3,6 Pode-se observar infiltrado linfo-histiocitário perivascular na derme papilar e pseudocistos córneos, com número de melanócitos normal.7,10 Testes genéticos revelam mutações em queratina 5 (KRT5), proteína O- glicosiltransferase-1 (POGLUT1), proteína O-fucosiltransferase-1 (POFUT1) e gene PSENEN como causadoras da doença, sendo que pacientes com esta última mutação apresentam lesões de hidradenite supurativa.2,4
Não há descrito nenhum tratamento definitivo para a DDD.7 Os tratamentos são de resultado insatisfatório, podendo ser empregados hidroquinona tópica, tretinoína, adapaleno e corticosteroides, além de laser Er:YAG, com o objetivo de reduzir o risco de hiperpigmentação pós-inflamatória.1,7 A isotretinoína seria uma opção, já que se trata de alteração da queratinização.1 Lesões císticas e tumorais devem ser tratadas com excisão cirúrgica.7
Foi realizado um estudo retrospectivo descritivo de dois casos atendidos na Clínica de Dermatologia com diagnóstico de doença de Dowling-Degos no período de novembro de 2016 a maio de 2019.
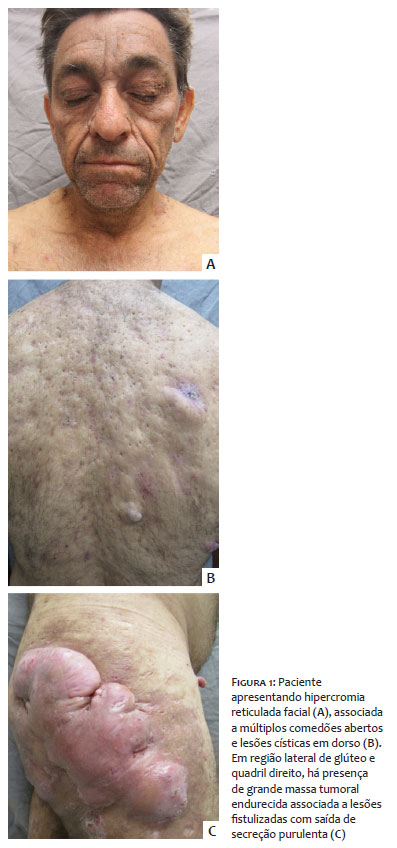
Homem de 58 anos, referindo hiperpigmentação localizada em face, região cervical e flexuras há dez anos, associada a aparecimento progressivo de múltiplos cistos, que drenavam exsudato purulento, e tumoração em nádega e face lateral de coxa direita. Ao exame clínico, apresentava em face e áreas flexurais hiperpigmentação predominante, múltiplos comedões abertos e cistos epidérmicos distribuídos pelo corpo, e em quadril e nádegas, lesões hidrosadenite-símile e tumoração de aspecto queloidiforme (Figura 1). Negava casos semelhantes na família.
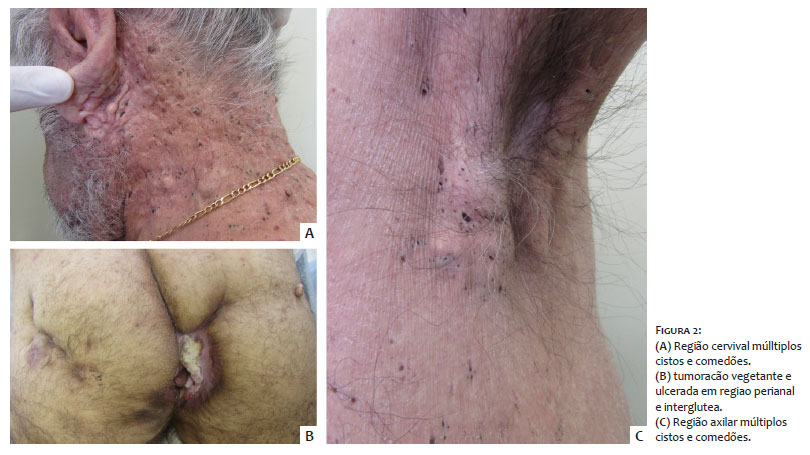
Homem de 59 anos, queixando-se de lesões supurativas na região glútea associadas à tumoração de crescimento progressivo há um ano. Ao exame clínico, apresentava dermatose caracterizada por múltiplos comedões disseminados pelo corpo, associados a lesões hidradenite-símile em glúteo esquerdo e tumoração vegetante e ulcerada em região perianal e interglútea, cuja biópsia diagnosticou carcinoma espinocelular invasivo (Figura 2). Negava casos semelhantes na família.
DDD é uma genodermatose de início tardio, em geral na idade adulta, que acomete inicialmente as axilas e virilhas, e mais tardiamente, as regiões interglútea e inframamária, pescoço e tronco.1 Ambos os pacientes apresentados tiveram o quadro iniciando-se na fase adulta e evidenciam claramente a predileção das lesões por estas localizações. Apesar de esta doença acometer predominantemente mulheres numa proporção de 2:11, apresentamos dois pacientes do sexo masculino.
Os pacientes aqui relatados apresentavam não só as manifestações discrômicas da síndrome, mas também diversas alterações da unidade pilossebácea disseminadas pelo corpo, de comedões abertos a hidradenite, com alto impacto na qualidade de vida em geral e autoestima em particular, inclusive pelas importantes alterações estéticas.
A exuberância das lesões aqui apresentadas demonstra o porquê da sinonímia de “doença dos pontos pretos” (dark dot disease – DDD) referida por alguns autores.1 Tais lesões, apesar de não apresentarem fenômenos inflamatórios na sua evolução benigna, são profundamente inestéticas. No segundo paciente foi, inclusive, evidenciado surgimento de carcinoma espinocelular interglúteo, neoplasia relatada na associação com esta doença.2,4 Além do aspecto clínico característico de ambos os casos, os diagnósticos foram confirmados com exame histopatológico que foi compatível aos achados descritos na literatura.
Por fim, é importante ressaltar que o presente estudo foi realizado na Clínica de Dermatologia de hospital terciário referência da maior cidade do país, e em três anos de atendimentos apenas dois casos de DDD foram evidenciados, mostrando a raridade desta dermatose na população geral.
Anna Carolina Brandão Vasconcelos | 000-0002-2171-4358
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
John Verrinder Veasey | 0000-0002-4256-5734
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
1. Zimmermann CC, Sforza D, Macedo PM, Azulay-Abulafia L, Alves MFGS, Carneiro SCS. Doença de Dowling-Degos: apresentação clínica e histopatológica clássica. An Bras Dermatol. 2011;86(5):979-82.
2. Fenske NA, Groover CE, Lober CW, Espinoza CG. Dowling-Degos disease, hidradenitis suppurativa, and multiple keratoacanthomas: a disorder that may be caused by a single underlying defect in pilsebaceous epithelial proliferation. J Am Acad Dermatol. 1991;24(5 Pt 2):888-92.
3. Kim YC, Davis MD, Schanbacher CF, Su WP. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999;40(3):462-7.
4. Linke M, Orouji A, Géraud C. Vesicular variant of Dowling-Degos disease. Br J Dermatol. 2018;179(3):795-6.
5. Ujihara M, Kamakura T, Ikeda M, Kodama H. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147(3):568-71.
6. Bhagwat PV, Tophakhane RS, Shashikumar BM, Noronha TM, Naidu V. Three cases of Dowling Degos disease in two families. Indian J Dermatol Venereol Leprol. 2009;75(4):398-400.
7. Hohmann CB, Koche B, Bonamigo RR, Dornelles ST, Cattani CAS. Caso para diagnostico. Doenca de Dowling-Degos e ceratoacantoma. An Bras Dermatol. 2010;85(2):241-3.
8. Gontijo B. O espectro doença de Kitamura - doença de Dowling-Degos. An Bras Dermatol. 1993;68(6):89-92.
9. Wu YH, Lin YC. Generalized Dowling-Degos disease. J Am Acad Dermatol. 2007;57(2):327-34.
10. Rathoriya SG, Soni SS, Asati D. Dowling-Degos disease with reticulate acropigmentation of Kitamura: extended spectrum of a single entity. Indian Dermatol Online J. 2016;7(1):32-5.
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}