


Cínthia Mendes1; Carolina Ferraz do Amaral1; Andre Luiz Simião2; Felipe Borba Calixto dos Santos3; Amilcar Castro4
The Merkel cell carcinoma is a rare tumor of neuroendocrine and epidermal origin and with poor prognosis. It is classically associated with immunosuppression, exposure to the sunlight and, more recently, with the polyomavirus. It is positive for epithelial and neuroendocrine markers. The combined expression of these markers confirms the diagnosis. Polyomavirus tumors have an unfavorable prognosis. The authors report a case of Merkel cell carcinoma with atypical immunophenotype (CK20 negative) and aggressive behavior. The present report is aimed at highlighting the importance of dermatologists having knowledge of different immunophenotypes that may be associated with the Merkel cell carcinoma.
Keywords: CARCINOMA, MERKEL CELL; IMMUNOHISTOCHEMISTRY; MERKEL CELL POLYOMAVIRUS
Initially described by Toker in 1972, Merkel cell carcinoma (MCC) is a rare and aggressive cutaneous neoplasm, with a slight preference for males and higher incidence of Caucasians at a mean age of 65 years at diagnosis.1 The most up to date evidence indicates that the neoplasia originates in cutaneous pluripotent stem cells, particularly those of epidermal lineage. This hypothesis supported by frequent association with other tumors originating in the epidermis, such as squamous cell carcinoma and Bowen’s disease.2
Classically, MCC is associated with chronic exposure to sunlight and immunosuppression.3,4 Transplanted patients, bearers of HIV infection and hematological malignancies constitute a risk group.
There are literature reports of regression of these tumors after reconstitution of the immune function in immunosuppressed patients, as well as descriptions of spontaneous regression, suggesting that prompt recognition of the lesions by the immune system may lead to the regression of the carcinoma.4
In 2008, a virus of the polyomavirus family (Merkel cell polyomavirus) was described, for which 80-90% of the CCMs cases were positive. Nevertheless, the real determinant of the oncogenic potential of this virus remains unclear.2,4 Cases of MCCs associated with polyomavirus (MCPyV+), however, seem to have a better prognosis and longer disease-free survival time, possibly due to the virus’ ability to stimulate the host’s immune response.4
The present article reports a case of MCC with atypical immunophenotype and aggressive behavior, which corroborates with current literature data, showing that these cases present worse development and prognosis.
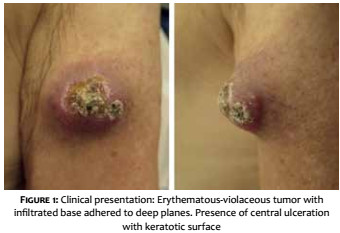
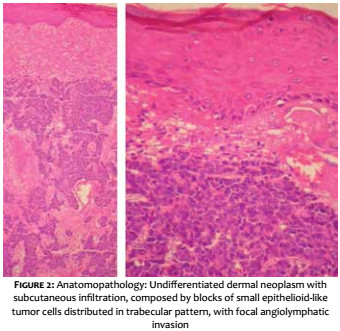
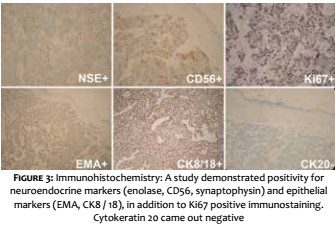
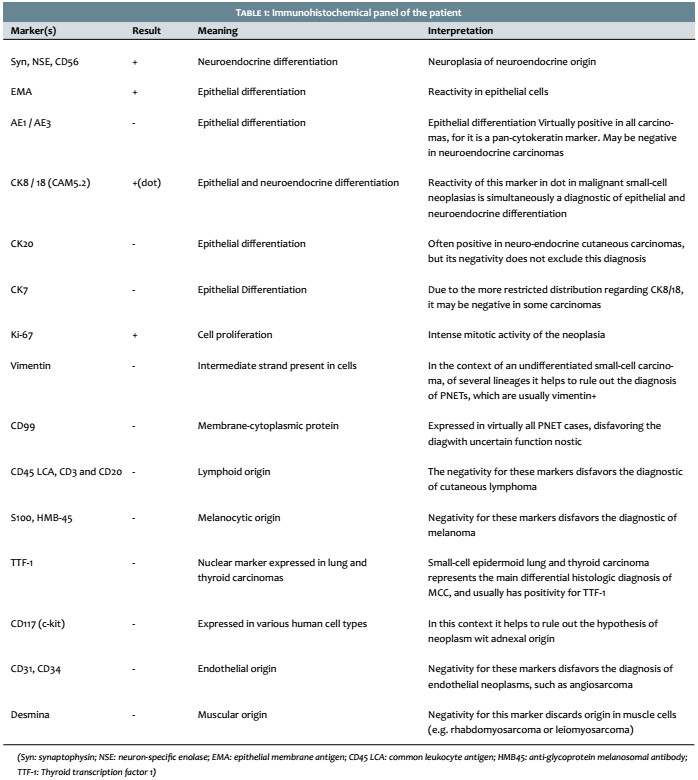
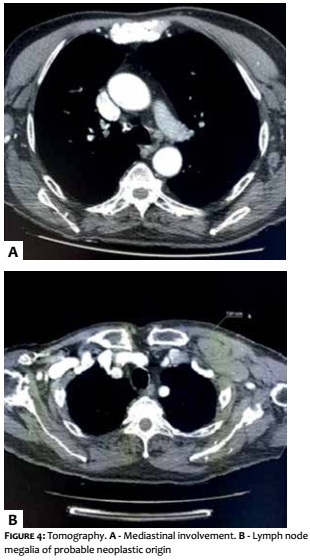
A 69-year-old white male patient reported an asymptomatic lesion of progressive growth in the left upper limb noticed six months before. He had systemic arterial hypertension, type 2 diabetes mellitus, heart failure, psoriasis and psoriatic arthritis. On physical examination, an erythematous-violaceous mass was observed on an infiltrate base, hardened and adhered to deep planes, of approximately 4 cm in diameter, with central ulceration surmounted by hyperkeratosis, on the anterior aspect of the left arm (Figure 1). There was absence of palpable lymph nodes. An excisional biopsy was performed, revealing an undifferentiated dermal neoplasm with subcutaneous infiltration, composed of blocks of small epithelioid tumor cells distributed in a trabecular pattern, with focal angiolymphatic invasion (Figure 2). The immunohistochemical study showed positivity for neuroendocrine markers (enolase, CD56 and synaptophysin) and epithelial markers (EMA and CK8/18), in addition to positive immunostaining for Ki-67, which indicates the intense mitotic activity of the neoplasia (Figure 3). The CK8/18 marker showed dot or perinuclear pattern reactivity, a fact that is representative of concomitant neuroendocrine and epithelial differentiation in a malignant small cell neoplasm. Immunostaining for cytokeratins 7 and 20 came out negative. The immunohistochemical panel is shown in Table 1. Excluding primary sites in other topographies, the final diagnosis was primary cutaneous neuroendocrine carcinoma, or MCC. Imaging studies were then performed for adequate staging and patient follow-up. Computerized tomography scans (CT) of the chest and abdomen showed images suggestive of secondary involvement in the mediastinum and liver (Figure 4a). Three months after the initial surgery, the patient presented a hardened mass of approximately 7cm in diameter in the infraclavicular region, on the left, which the CT examination evidenced as a lymph node megalia of probable metastatic origin (Figure 4b). In addition, considerable clinical signs and symptoms of consumptive disease, such as anorexia and weight loss, were observed. The patient died 3 months later, despite having been treated with systemic chemotherapy.
Clinically, MCC emerges as a solitary nodule or erythematous or violaceous plaque, of firm and rapid growth, usually painless, with eventual ulceration, in the head or neck regions. Trunk, extremities and photoprotected areas are less frequent locations. Due to the lack of specificity of its clinical appearance, Heath et al. proposed the AEIOU acronym, in an attempt to aid diagnosis (Asymptomatic, Expanding rapidly, Immune suppression, Older than 50, Ultraviolet exposed site).1 Local recurrence is very frequent, there is locoregional involvement in 17% to 76% of cases, and distant metastases occurs in approximately 50%, both hematogenously and lymphatically, with a lethality rate ranging from 20% to 55%. The most affected organs are (starting with the highest frequency): skin, lymph nodes, liver and lungs. Five-year survival rate is of 64% for localized disease, 39% for lymph node involvement, and 18% for distant metastases4
Histology evidences the neoplasia as an poorly-defined dermal nodule that frequently infiltrates fat, fascia, and muscle.5 The cell infiltrate is uniform and monotonous, composed of small round oval basal cells with vesicular ovoid nucleus and non-prominent nucleolus, in addition to scarce cytoplasm with numerous mitotic figures and apoptotic bodies.6,7 Areas of extensive or focal necrosis are common. Three variants are described: trabecular, intermediate and small-cell, nonetheless mixed or transitional forms are more commonly found.3 The relationship between histologic type and prognosis is controversial. Histological findings are not characteristic and the main differential diagnoses include cutaneous metastasis from small-cell lung carcinoma, cutaneous lymphoma, melanoma, primitive neuroectodermal tumors (PNET), and squamous cell carcinoma (SCC). It is worth noting that, not infrequently, MCC occurs concomitantly with other lesions of epithelial origin, the most common being the association with invasive SCC. Bowen’s disease, basal cell carcinoma (BCC), actinic keratosis and sebaceous carcinoma have also been reported.2,5,6
After the histological analysis, the immunohistochemical study becomes mandatory for diagnostic definition. Characteristically, MCC is positive for epithelial markers, such as cytokeratins 20 and 8/18, and neuroendocrine markers, such as chromogranin (CgA), synaptophysin (Syn) and neurospecific enolase (NSE). The combined expression of these markers is corroborated by the diagnosis.1-7 Cytokeratin 20 is considered a standard marker in cases of MCC, since it is present in up to 95% of cases, often expressed as a perinuclear dot pattern.2,3,4 CK20 expression is absent in most small-cell and round-cell neoplasms, except for MCC.
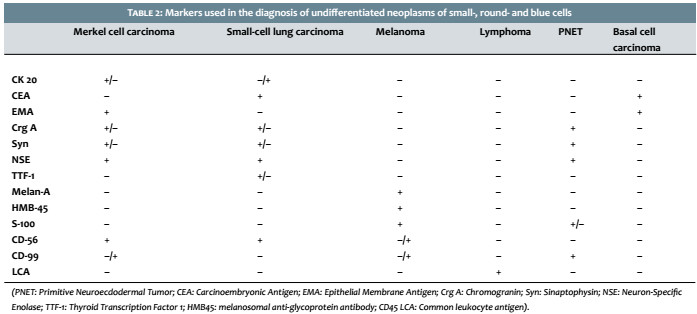
However, the absence of reactivity for CK20 does not exclude MCC as a diagnostic possibility. In these cases, it is necessary to analyze other cytokeratins such as AE1 / AE3 (pan-cytokeratin), CK8/18 or Cam5.2 and CK7, in addition to verifying the expression of other markers that are usually negative in Merkel cases. These markers are: thyroid transcription factor 1 (TTF-1), Melan-A, HMB45 (Human Melanoma Black), S-100, common leukocyte antigen (CLA), and CD99 .2-5,8 Table 2 depicts the main markers that should be evaluated after the histological diagnosis of small-cell, round-cell a Table 2).
The typical immunostaining of MCC is performed with CK20+ and CK7-, nevertheless any combination of the expression of these cytokeratins can be found (CK20+ / CK7+, CK20+ / CK7-, CK20- / CK7-, CK20- / CK7+) and the actual significance of these immunophenotypes still needs to be determined. In 2013, Ishida and Okabe reported 2 cases of MCC associated with Bowen’s disease, one of which had a rather infrequent immunophenotype (CK20e TTF-1+). These authors concluded that collision tumors may have unusual immunophenotypes, and that atypical immunohistochemical patterns generally do not involve infection detectable by MCPyV.2
The breakthrough for the understanding of part of the MCC’s pathogenesis took place in 2008 after the discovery of a polyomavirus, termed Merkel cell polyomavirus, for which positivity is observed in 80-90% of the Merkel tumors.2,4,9 The polyomavirus could promote tumorigenesis through the oncogenic action of small and large T antigens (LT [large] and ST [small] -Ag) with subsequent integration of the viral genome into the host, which seems to occur early after infection by MCPyV.4,8,9 Since its original description in 2008, epidemiological data have strongly supported the virus’ correlation to MCC.
However, the real determinant of this virus’ oncogenic potential remains unclear. Roughly 60%-80% of the normal population is positive for MCPyV infection, nevertheless only a minority develops the neoplasia.9 A reported finding is that patients infected with MCC have much higher levels of antibody to the virus than infected patients without the disease.9 However, MCC cases associated with polyomavirus appear to have a better prognosis and longer disease-free survival, possibly related to the ability of the virus to stimulate the host’s immune response.4,9 It is questioned whether immunosuppression would be the predisposing factor for the development of MCC in patients infected with the virus, since the neoplasia is much more frequent in this population. Notwithstanding, there is absence of studies demonstrating that MCCs with MCPyV positivity are more common in immunocompromised individuals.
It is believed that exposure to the virus and the resulting infection occurs in early childhood, being, however, clinically asymptomatic due to the fact it produces adequate humoral and cellular responses. Ultraviolet radiation and other potentially mutagenic environmental factors would be responsible for the integration of the viral genome into host DNA, with subsequent development of the neoplasm in adulthood. Concomitantly, systemic immunodepression, local or even induced by the own tumor would contribute to tumor proliferation. The disease’s progression can be monitored by anti-T-Ag antibody levels and the outcome of the picture can be predicted by LTCD8+ levels in the tumor infiltrate (high levels of LTCD8+ correlate with a better prognosis). MCPyV tumors often associate with aggressive somatic mutations (RB1,Tp53 and PIK3CA) and have an unfavorable prognosis, as they probably develop via a different oncogenic pathway. Knowledge of the biological behavior of polyomavirus-positive tumors seems to be considerably promising for the development of therapies specifically focusing on tumor-related proliferation targets.4,8
In 2015, Miner et al., from the University of Michigan, questioned the association of negativity to CK20 with the absence of polyomavirus infection, finding 10 cases without MCPyV positivity among the 13 studied CK20- (77%). Therefore, it was concluded that the CK20 MCCs are associated with a lower incidence of MCPyV positivity. Further studies are needed to establish whether the CK20 MCPyV MCC is genetically similar to other CK20 CCM, but MCPyV+ or whether this tumor subgroup has a single spectrum of mutations and would be a distinct class of CCM8 (Table 3).
Treatment is based on complete surgical excision associated with adjuvant treatments, such as chemo and radiotherapy, depending on the extent of the disease. Postoperative radiotherapy of the tumor bed and regional lymph nodes is advocated aimed at better locally controlling the condition, due to the tumor’s radiosensitivity, and lower recurrence rates. It presents proven and consensual application and benefits also in recurrent or unresectable tumors. Chemotherapy is a palliative option in advanced stages, with a positive response in two thirds of patients, however with recurrence within a few months. The proposed macroscopic margins range from 1cm to 3cm and the Mohs technique is well indicated for locations where this extension of margins may be impractical, such as in the face. Nonetheless, there remain controversies regarding the best therapeutic approach. Considering that there is subclinical lymph node disease in 25%-50% of cases, sentinel lymph node research is recommended. Moreover, lymphadenectomy is indicated in case of presence of clinical or histological lymph node involvement.3
The disease’s prognosis is not good due to the high rates of local recurrence, lymph node and distant metastasis. The average 5 years survival rate is of 30%-75%, and usually ranges from 6 to 12 months. The factors most frequently associated with a worse prognosis include: male gender, large primary tumor, presence of lymph node or distant metastases at diagnosis, histological evidence of nuclear atypia, increased cell turnover and angiolymphatic invasion, MCPyV negativity, somatic mutations associated with CK20 (for instance Tp53 or PIK3CA), increased expression of markers such as Ki-67, and poor expression of other markers, such as CD34.4
1. Mello DF, Ricciluca L, Felix M, Rodrigues A, Helene Jr A. Carcinoma das células de Merkel: relato de 2 casos. Rev Bras Cir Plást. 2010(25): 217-21.
2. Ishida M, Okabe H. Merkel cell carcinoma concurrent with Bowen's disease: two cases, one with an unusual immunophenotype. J Cutan Pathol. 2013(40): 839-43.
3. Duprat JP, Landman G, Salvajoli JV, Brechtbühl ER. A Review of the epidemiology and treatment of Merkel cell carcinoma. Clinics 2011;66(10):1817-1823.
4. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel Cell Carcinoma: Implications for Immunotherapy of Polyomavirus-Associated Cancer. Curr Oncol Rev. 2011.
5. He W, Zhang D, Jiang J, Chen Y, Wu C. Merkell cell carcinoma in the left groin: A case report and review of the literature. Oncol Lett. 2015(9): 1197-1200.
6. Rossoe EWT, Fernandes KKML, Prado IDF, Bazzo ILMS, Tebcherani AJ, Santos TC. Tumor de Merkel: relato de caso. Surg Cosmet Dermatol. 2012(4):268-70.
7. Pilloni L, Manieli C, Senes G, Ribuffo D, Faa G. Merkel cell carcinoma with an unusual immunohostochemical profile. Eur J Histochem. 2009(53): 275-8
8. Miner AG, Patel RM, Wilson DA, Procop GW, Minca EC, Fullen DR, et al. Cytokeratin 20-negative Merkel cell carcinoma is in frequently associated with the Merkel cell polyomavirus. Mod Pathol. 2015(28):498-504.
9. Erstad DJ, Cusack Jr JC. Mutational Analysis of Merkel Cell Carcinoma. Cancers. 2014(6): 2116-36.
This study was carried out at the Hospital and Maternity Celso Pierro, Pontifícia Universidade Católica de Campinas (PUCCAMP) - Campinas (SP), Brazil.
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}