


Cínthia Mendes1; Carolina Ferraz do Amaral1; Andre Luiz Simião2; Felipe Borba Calixto dos Santos3; Amilcar Castro4
O carcinoma de células de Merkel (CCM) é um tumor raro de origem neuroendócrina e epidérmica, de mau prognóstico. Está classicamente associado à imunossupressão, exposição solar e, mais recentemente, ao poliomavírus (MCPyV). Caracteristicamente, o carcinoma de células de Merkel apresenta positividade para marcadores epiteliais e neuroendócrinos. A expressão combinada desses marcadores é o dado que corrobora o diagnóstico. Tumores MCPyV- possuem prognóstico desfavorável. Relata-se um caso de carcinoma de células de Merkel com imunofenótipo atípico (CK20 negativo) e comportamento agressivo. Este relato se justifica para reforçar a importância do conhecimento, pelos dermatologistas, de diferentes imunofenótipos que podem estar associados ao carcinoma de células de Merkel.
Keywords: CARCINOMA DE CÉLULA DE MERKEL; IMUNO-HISTOQUÍMICA; MERKEL CELL POLYOMAVIRUS
O carcinoma de Células de Merkel (CCM), descrito inicialmente por Toker, em 1972, é uma neoplasia cutânea rara e agressiva, com discreta predileção para o sexo masculino, que incide mais frequentemente em caucasianos com idade média de 65 anos ao diagnóstico.1 As evidências mais atuais apontam que a neoplasia tem origem em células-tronco pluripotentes cutâneas, particularmente da linhagem epidérmica; hipótese apoiada pela frequente associação com outros tumores originados na epiderme, como carcinoma espinocelular e doença de Bowen.2
Classicamente, o CCM está associado à exposição solar crônica e imunossupressão.3,4 Pacientes transplantados, com infecção pelo HIV (Vírus da Imunodeficiência Humana) e portadores de neoplasias hematológicas constituem grupo de risco. Há na literatura relatos de regressão desses tumores após reconstituição da função imune em pacientes imunossuprimidos, bem como relatos de regressão espontânea, sugerindo que um pronto reconhecimento da lesão pelo sistema imune possa levar ao "clareamento" do carcinoma.4
Em 2008 foi descrito um vírus da família poliomavírus (Merkel cell polyomavirus), para o qual se demonstra positividade em 80-90% dos CCM, contudo, o real fator determinante do potencial oncogênico deste vírus permanece não esclarecido.2,4 Os casos de CCM associados ao poliomavírus (MCPyV+), no entanto, parecem apresentar melhor prognóstico e maior tempo de sobrevida livre de doença, provavelmente relacionado a capacidade do vírus em estimular a resposta imunológica do hospedeiro.4
Relata-se um caso de carcinoma de células de Merkel com imunofenótipo atípico e comportamento agressivo, o que corrobora com os dados atuais da literatura que demonstram que tais casos apresentam pior evolução e prognóstico.
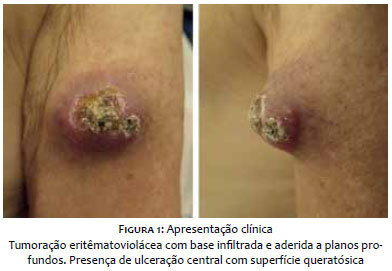
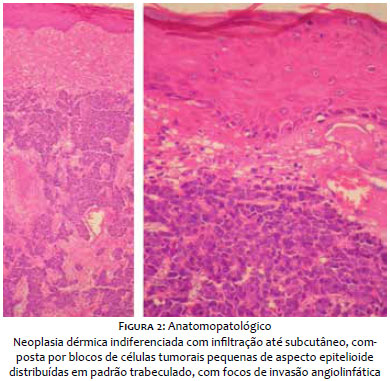
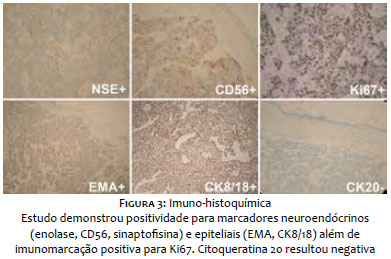
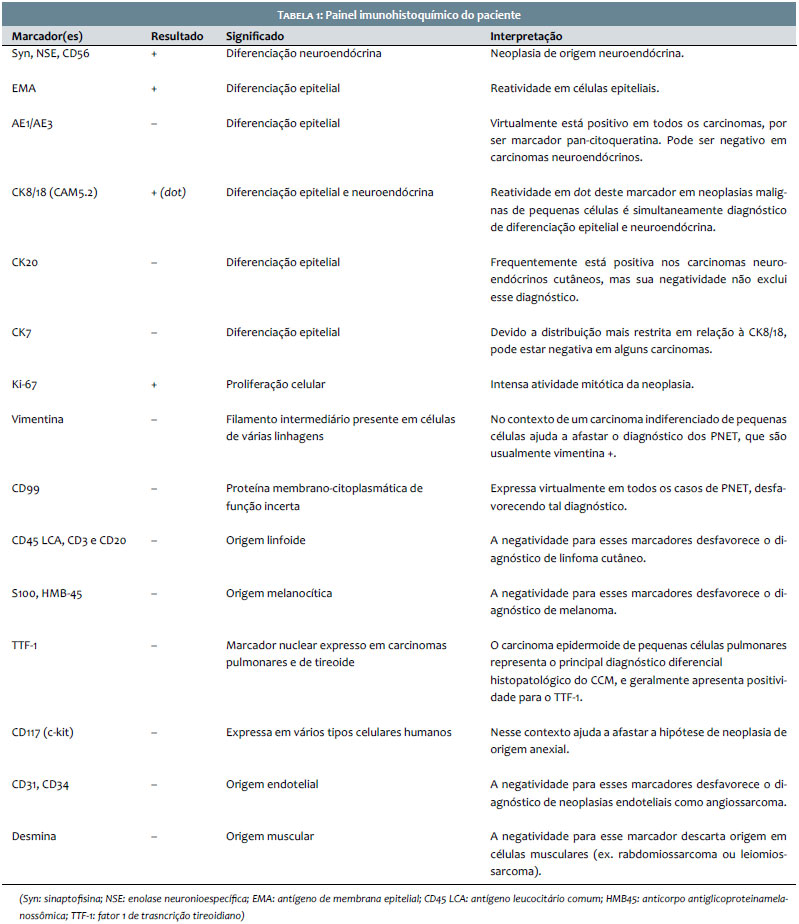
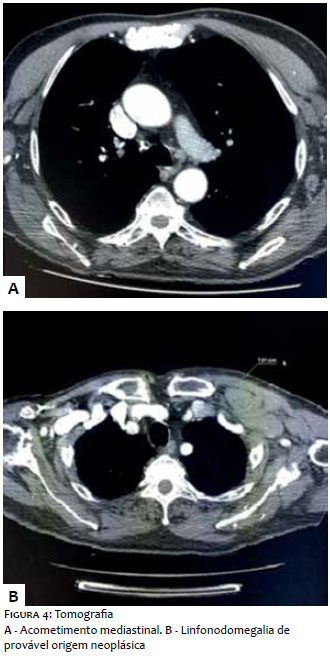
Paciente do sexo masculino, branco, 69 anos, relatava lesão assintomática de crescimento progressivo no membro superior esquerdo há seis meses. Era portador de hipertensão arterial sistêmica, diabete melito tipo 2, insuficiência cardíaca, psoríase e artrite psoriática. Ao exame físico observava-se tumoração eritêmato-violácea sobre base infiltrada, endurecida e aderida a planos profundos, de aproximadamente 4 cm de diâmetro, com ulceração central encimada por hiperceratose, na face anterior do braço esquerdo (Figura 1). Ausência de linfonodos palpáveis. Realizada biópsia excisional cujo estudo anátomo-patológico evidenciou neoplasia dérmica indiferenciada com infiltração até o subcutâneo, composta por blocos de células tumorais pequenas de aspecto epitelioide distribuídas em padrão trabeculado, com focos de invasão angiolinfática (Figura 2). Seguiu-se o estudo imunohistoquímico que demonstrou positividade para marcadores neuroendócrinos (enolase, CD56 e sinaptofisina) e epiteliais (EMA e CK8/18), além de imunomarcação positiva para Ki-67, o que denota a intensa atividade mitótica da neoplasia (Figura 3). O marcador CK8/18 apresentava reatividade em padrão dot ou perinuclear, fato este representativo de diferenciação neuroendócrina e epitelial concomitantes em uma neoplasia maligna de pequenas células. A imunomarcação para citoqueratinas 7 e 20 resultou negativa. O painel imunohistoquímico está demonstrado na tabela 1. Excluídos sítios primários em outras topografias, o diagnóstico final foi o de carcinoma neuroendócrino cutâneo primário, ou CCM. Foram então realizados estudos de imagem para adequados estadiamento e seguimento do paciente. As tomografias de tórax e abdome demonstraram imagens sugestivas de acometimento secundário no mediastino e no fígado (Figura 4a). Após três meses da cirurgia inicial o paciente apresentou massa endurecida de aproximadamente 7 cm de diâmetro na região infraclavicular à esquerda, que a tomografia mostrou tratar-se de linfonodomegalia de provável origem metastática (Figura 4b), além de consideráveis sinais e sintomas clínicos de doença consumptiva como anorexia e emagrecimento. O paciente foi a óbito 3 meses após, apesar de terapia sistêmica quimioterápica.
Clinicamente, o CCM apresenta-se como nódulo solitário ou placa eritematosa ou violácea, firme, de crescimento rápido, geralmente indolor, com eventual ulceração, na cabeça ou no pescoço. Tronco, extremidades e áreas fotoprotegidas são localizações de menor frequência. Devido à inespecifidade de sua apresentação clínica, Heath e cols. propuseram o acrômio AEIOU, na tentativa de auxiliar o diagnóstico (Asymptomatic, Expanding rapidly, Immune supression, Older than 50 years, Ultraviolet exposed site).1 A recorrência local é muito frequente, há acometimento locorregional em 17 a 76% dos casos e metástases à distância ocorrem em aproximadamente 50%, tanto por via hematogênica quanto linfática, com taxa de letalidade que varia de 20 a 55%. Os órgãos mais acometidos são, em ordem de frequência, pele, linfonodos, fígado e pulmões. A sobrevida em 5 anos é de 64% para doença localizada, 39% para acometimento linfonodal e 18% para metástases à distância.4
Na histopatologia, a neoplasia mostra-se como um nódulo dérmico, mal definido, que frequentemente infiltra a gordura, fáscia e músculo.5 O infiltrado celular é uniforme e monótono composto de pequenas células basalóides redondas a ovais, com núcleo ovoide vesicular e nucléolo não proeminente, e citoplasma escasso, com numerosas figuras de mitose e corpos apoptóticos.6,7 Áreas de necrose extensa ou focal são comuns. São descritas três variantes: trabecular, intermediária e de pequenas células, no entanto, formas mistas ou transicionais são as mais comumente encontradas.3 A relação entre o tipo histológico e o prognóstico é controversa. Os achados histopatológicos não são característicos e os principais diagnósticos diferenciais incluem: metástase cutânea de carcinoma de pequenas células do pulmão, linfoma cutâneo, melanoma, tumores neuroectodérmicos primitivos (PNET) e carcinoma espinocelular (CEC) pouco diferenciado.1,5,7 É importante ressaltar que, não infrequentemente, o CCM ocorre de maneira concomitante a outras lesões de origem epitelial, sendo mais comum a associação com CEC invasivo. Doença de Bowen, carcinoma basocelular, queratose actínica e carcinoma sebáceo também já foram associações relatadas.2,5,6
Após a análise histopatológica, o estudo imunohistoquímico se faz mandatório para a definição diagnóstica. Caracteristicamente, o CCM apresenta positividade para marcadores epiteliais, como citoqueratinas 20 e 8/18, e neuroendócrinos, como cromogranina (CgA), sinaptofisina (Syn) e enolase neuroespecífica (NSE). A expressão combinada desses marcadores é o dado que corrobora o diagnóstico.1,2,3,4,5,6,7 A citoqueratina 20 é considerada um marcador padrão nos casos de CCM, já que está presente em até 95% dos casos, frequentemente expressa com padrão em dot perinuclear.2,3,4 A expressão de CK20 está ausente na maior parte das neoplasias de células pequenas e redondas, à exceção do CCM.
No entanto, a ausência de reatividade para CK20 não exclui o CCM como possibilidade diagnóstica, sendo necessário, nesses casos, a análise de outras citoqueratinas como AE1/AE3 (pan-citoqueratina), CK8/18 ou Cam5.2 e CK7, além do estudo da expressão de outros marcadores que usualmente estão negativos nos casos de Merkel, como o fator 1 de transcrição da tireoide (TTF-1), Melan-A, HMB45 (Human Melanoma Black), S-100, antígeno leucocitário comum (LCA) e CD99.2,3,4,5,8 A tabela 2 apresenta os principais marcadores que devem ser avaliados após o diagnóstico histopatológico de uma neoplasia de células pequenas, redondas e azuis pequenas, redondas e azuis. (Tabela 2).
A imunomarcação típica do CCM é CK20+ e CK7-, no entanto, qualquer combinação da expressão dessas citoqueratinas é possível de ser encontrada (CK20+/CK7+, CK20+/CK7-, CK20-/CK7-, CK20-/CK7+) e a real significância desses imunofenótipos ainda precisa ser determinada. Em 2013, Ishida e Okabe relataram 2 casos de CCM associados à doença de Bowen, sendo que um deles, apresentava um imunofenótipo bastante infrequente (CK20- e TTF-1+). Esses autores concluíram que tumores de colisão podem apresentar imunofenótipos não usuais e, padrões atípicos de imunohistoquímica geralmente não envolvem infecção detectável pelo MCPyV.2
A revolução para o entendimento de parte da patogenia do CCM ocorreu em 2008 após a descoberta de um poliomavírus, denominado Merkel cell polyomavirus, para o qual se observa positividade em 80-90% dos tumores de Merkel.2,4,9 O poliomavírus poderia promover a tumorigênese através da ação oncogênica de antígenos T pequenos e grandes (LT[large] e ST [small]-Ag) com posterior integração do genoma viral ao do hospedeiro, o que parece ocorrer precocemente após a infecção pelo MCPyV.4,8,9 Desde sua descrição original em 2008, os dados epidemiológicos tem apoiado fortemente a relação do vírus com o CCM.
Contudo, o real fator determinante do potencial oncogênico deste vírus permanece não esclarecido. Em torno de 60 a 80% da população normal apresenta positividade para infecção pelo MCPyV, no entanto, apenas uma minoria desenvolve a neoplasia.9 Um dado relatado é que os pacientes infectados com CCM apresentam níveis de anticorpos contra o vírus muito mais altos que os pacientes infectados sem a doença.9 Os casos de CCM associados ao poliomavírus, no entanto, parecem apresentar melhor prognóstico e maior tempo de sobrevida livre de doença, provavelmente relacionados à capacidade do vírus em estimular a resposta imunológica do hospedeiro.4,9 É questionado se a imunodepressão seria o fator predisponente para o desenvolvimento do CCM nos pacientes infectados pelo vírus, já que a neoplasia é muito mais frequente nesta população, no entanto, não existem estudos que demonstrem que os CCM com positividade para o MCPyV sejam mais comuns em imunodeprimidos.
Acredita-se que a exposição ao vírus e consequente infecção ocorre na infância precoce, no entanto, ela é clinicamente assintomática, pois produz resposta humoral e celular adequadas. A radiação ultravioleta e outros fatores ambientais potencialmente mutagênicos seriam responsáveis pela integração do genoma viral ao DNA do hospedeiro com posterior desenvolvimento da neoplasia, na idade adulta. Em paralelo, imunodepressão sistêmica, local ou mesmo induzida pelo próprio tumor contribuiriam para a proliferação tumoral. A progressão da doença pode ser monitorada pelos níveis de anticorpos anti-T-Ag e o desfecho do quadro pode ser predito pelos níveis de LTCD8+ no infiltrado tumoral (níveis altos de LTCD8+ correlacionam-se com melhor prognóstico). Tumores MCPyV- frequentemente associam-se a mutações somáticas agressivas (RB1, Tp53 e PIK3CA) e possuem prognóstico desfavorável, pois provavelmente desenvolvem-se através de uma via oncogênica diferente. O conhecimento do comportamento biológico dos tumores positivos para o poliomavírus parece ser bastante promissor quanto ao desenvolvimento de terapêuticas que atuem especificamente contra os alvos relacionados à proliferação tumoral.4,8
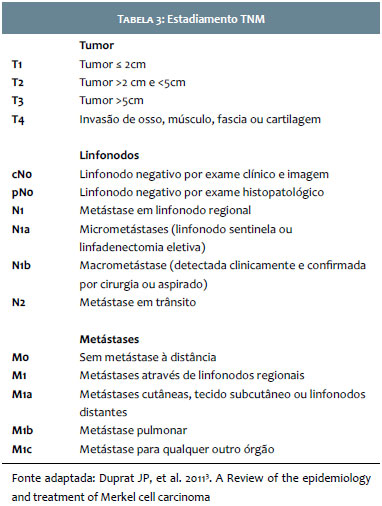
Em 2015, Miner e colaboradores, da Universidade de Michigan, questionaram a associação da negatividade para a CK20 com a ausência de infecção para o poliomavírus e encontraram 10 casos sem positividade para o MCPyV entre os 13 estudados CK20-(77%). Concluiu-se, portanto, que os CCM CK20- estão associados a menor incidência de positividade para o MCPyV. Mais estudos são necessários para estabelecer se o CCM CK20- MCPyV- é semelhante geneticamente a outros CCM CK20- porém MCPyV+ ou se esse subgrupo de tumores possui espectro único de mutações e seriam uma classe distinta de CCM8 (Tabela 3).
O tratamento é baseado na exérese cirúrgica completa associada ou não a tratamentos adjuvantes, como quimio e radioterapia, a depender da extensão da doença. Radioterapia pós-operatória do leito tumoral e da cadeia linfática é defendida considerando melhor controle local, devido à radiossensibilidade do tumor, e menores índices de recidiva. Ela apresenta aplicação e benefícios comprovados e consensuais também em tumores recidivantes ou irressecáveis. A quimioterapia é uma opção paliativa nos estádios avançados, com resposta positiva em dois terços dos pacientes, porém frequente recorrência em alguns meses. As margens macroscópicas propostas variam de 1 a 3 cm e a técnica de Mohs está bem indicada para locais nas quais elas possam ser impraticáveis como, por exemplo, a face. No entanto, ainda permanecem controvérsias em relação a melhor abordagem terapêutica. Considerando-se que há doença linfonodal subclínica em 25-50% dos casos é preconizada a pesquisa de linfonodo sentinela, e na presença de acometimento linfonodal, clínica ou histologicamente, está indicada a linfadenectomia.3
O prognóstico da doença não é bom, devido aos altos índices de recidiva local, metástases linfonodais e à distância. A sobrevida média em cinco anos é de 30-75%, e geralmente varia entre 6 e 12 meses. Os fatores mais associados a pior prognóstico incluem sexo masculino, tumor primário grande, presença, ao diagnóstico, de metástases linfonodais ou à distância, evidências histológicas de atipias nucleares, turnover celular aumentado e invasão angiolinfática, negatividade para o MCPyV, mutações somáticas associadas a CK20- (ex, Tp53 ou PIK3CA), expressão aumentada de marcadores como o Ki-67 e pobre de outros, como o CD34.4
1. Mello DF, Ricciluca L, Felix M, Rodrigues A, Helene Jr A. Carcinoma das células de Merkel: relato de 2 casos. Rev Bras Cir Plást. 2010(25): 217-21.
2. Ishida M, Okabe H. Merkel cell carcinoma concurrent with Bowen's disease: two cases, one with an unusual immunophenotype. J Cutan Pathol. 2013(40): 839-43.
3. Duprat JP, Landman G, Salvajoli JV, Brechtbühl ER. A Review of the epidemiology and treatment of Merkel cell carcinoma. Clinics 2011;66(10):1817-1823.
4. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel Cell Carcinoma: Implications for Immunotherapy of Polyomavirus-Associated Cancer. Curr Oncol Rev. 2011.
5. He W, Zhang D, Jiang J, Chen Y, Wu C. Merkell cell carcinoma in the left groin: A case report and review of the literature. Oncol Lett. 2015(9): 1197-1200.
6. Rossoe EWT, Fernandes KKML, Prado IDF, Bazzo ILMS, Tebcherani AJ, Santos TC. Tumor de Merkel: relato de caso. Surg Cosmet Dermatol. 2012(4):268-70.
7. Pilloni L, Manieli C, Senes G, Ribuffo D, Faa G. Merkel cell carcinoma with an unusual immunohostochemical profile. Eur J Histochem. 2009(53): 275-8
8. Miner AG, Patel RM, Wilson DA, Procop GW, Minca EC, Fullen DR, et al. Cytokeratin 20-negative Merkel cell carcinoma is in frequently associated with the Merkel cell polyomavirus. Mod Pathol. 2015(28):498-504.
9. Erstad DJ, Cusack Jr JC. Mutational Analysis of Merkel Cell Carcinoma. Cancers. 2014(6): 2116-36.
Trabalho realizado no Hospital da PUC Campinas - Maternidade Celso Pierro – Campinas (SP), Brasil
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}