


Tânia Rita Moreno de Oliveira Fernandes1; Graziele Áquila de Souza Brandão2; Talita Suzany Siqueira dos Santos3
Keywords: HISTIOCYTOMA, BENIGN FIBROUS; LUPUS ERYTHEMATOSUS, SYSTEMIC; IMMUNOSUPPRESSIVE AGENTS.
Solitary dermatofibromas or superficial benign fibrous histiocytomas are common and benign dermal fibrohistiocytic tumors that have the appearance of papules or nodules of a brownish erythematous color. Appearing as a single tumor, or in small numbers, they are generally asymptomatic. The diagnosis is clinical and histologic, and surgical treatment is recommended only for aesthetic reasons and if there are any symptoms - which is unusual. 1 Its etiology is unknown, and it is usually located in the lower limbs, with an absence of association with systemic diseases.
Multiple eruptive dermatofibromas (MED) constitute a variant, which occurs in 0.3% of patients, with the presence of 15 or more lesions, or the appearance of 5 to 8 tumors in less than four months. 1 The lesions are usually painful and widespread, and have been associated with autoimmune diseases, immunosuppressive drugs, and hormonal alterations. 2 Many patients have alterations in their immune status -more commonly AIDS and systemic lupus erythematosus (SLE) 3,4 - nevertheless other immune disorders (dermatomyositis, Sjögren's syndrome, hepatitis C) or myeloprolifarerative disorders (cutaneous t-cell lymphoma) can be associated with MED. 4 Furthermore, antiretroviral agents, immunobiological drugs (Efalizumab), and anti-tumor necrosis factor alpha may also be involved. Therefore, the emergence of this type of entity should prompt the investigation of the underlying disease. Various histological subtypes of dermatofibromas have been described in the scientific literature, however in general MED arises histologically as poorly circumscribed lesions, exhibiting epidermal hyperplasia, prominent collagen bundles and diffuse proliferation of fibrocytes. 5 The present report describes a case in which MED developed 20 years before the diagnosis of SLE.
A 47-year-old mulatto female patient, born and raised in the city of Juazeiro, Bahia State, Brazil, describe the emergence of approximately 5 nodular erythematous-brownish lesions on the lower limbs (LL), 25 years earlier, denying a history of local or systemic symptoms or medication use. The gradual emergence of other lesions with the same feature in the LL, upper limbs (UL) and abdomen ensued. New lesions have continued to appear up until the present time.
For the previous 5 years, the patient had been having protracted courses of fever for about 60 days, polyarthralgia, swelling in joints of the hands and feet, myalgia, loss of appetite, chest pain, and diffuse alopecia, which led her to seek care at the Rheumatology Department.
Laboratory tests revealed: ANF 1:1,500 (nuclear fine speckled pattern); hemangioma (Hb 10.7, leucocytes 8,000/ml, platelets 435,000; alpha-1 acid glycoprotein 241, N < 117; alpha-1 antitrypsin 282, N < 174); native anti-DNA 1:10, N = non-reactive; ESR 52mm in the first hour; echocardiography: limited pericardial effusion with cardiac chambers and normal diastolic function; normal renal and liver functions; negative PPD.
Once SLE was diagnosed, the patient was treated with 15 mg/day meloxicam, 30 mg/day famotidine, 200mg/day chloroquine diphosphate, 20mg/day prednisone and 10mg/week methotrexate, progressing to resolution of the fever and a progressive improvement in symptoms.
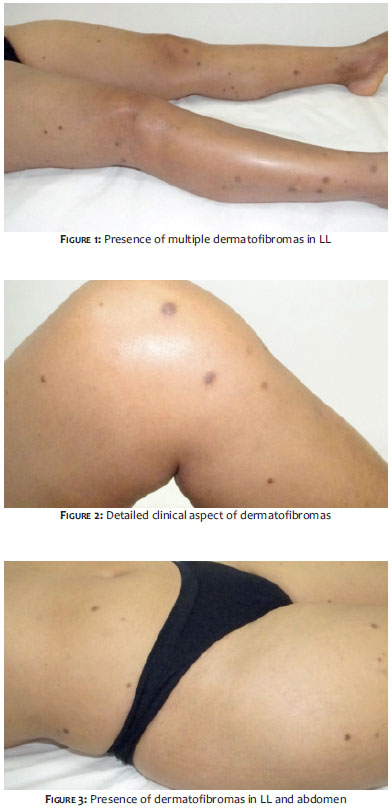
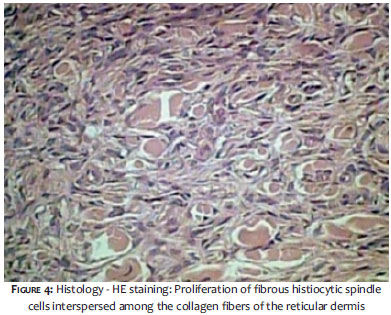
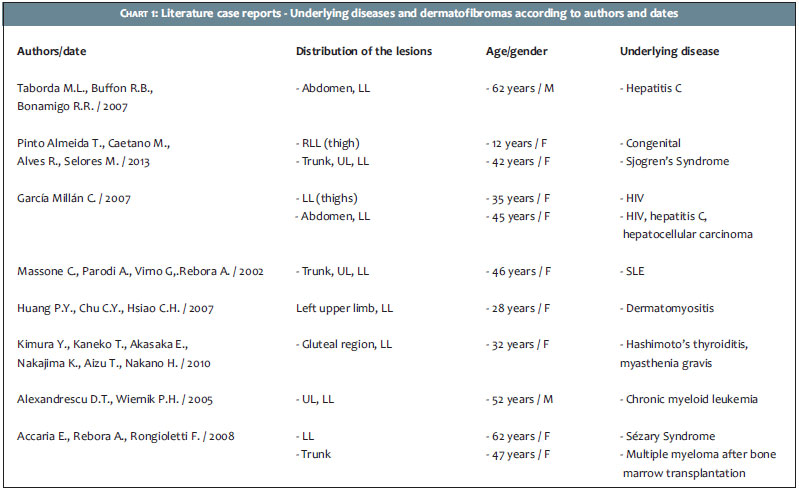
Two years after the onset of the systemic picture, the patient sought out the Dermatology Service due to the fact that the lesion presented a significant increase of brownish nodules, then numbering greater than 40, predominantly in the LL, UL, and abdomen (Figures 1 to 3). The biopsy of one of the lesions characterized the presence of dermatofibroma (Figure 4).
The patient had FAN 1:1,280 on the occasion, normal protein electrophoresis and echocardiogram, and was taking meloxicam, famotidine and 6mg/day deflazacort.
The patient has been followed up with by the Rheumatology and Dermatology departments to date, showing decreased ANF (1:320), thus confirming the control of the systemic involvement. She has, however, a progressive increase of the MED. After having been made aware of the benignity of the tumor, the patient chose not to undergo exeresis.
MED has been associated with several comorbidities (56%) 4 and autoimmune diseases treated with immunosuppressants - especially SLE (46%), HIV infection, ulcerative colitis, pemphigus vulgaris, acute myeloid leukemia, and organ transplants, and also congenital and family cases. 5 It occurs more frequently in female patients, which may be explained by the occurrence of autoimmune diseases being more predominant in women, particularly SLE.
Histologically it is characterized by proliferation in the dermis of non-encapsulated spindle cells with the periphery composed of bundles of collagen. The overlying epidermis is acanthotic and has hyperpigmentation in the basal layer. The presence of lymphocytic inflammatory infiltrate is common. 6
In the present case, the patient was diagnosed with MED 20 years after its onset and, in the course of this, was diagnosed with SLE due to the presence of non-scarring alopecia, arthritis in two or more peripheral joints, serositis, positive for FAN and anti-DNA. In this manner, the patient therefore met 5 of the criteria proposed by the Systemic Lupus International Collaborating Clinics, which set new criteria for SLE in 2012, basing the diagnosis on the presence of 4 of the 17 listed, with at least 1 clinical and 1 immune criterion, or a renal biopsy consistent with nephritis lupica associated with positive ANF or anti-DNA. 7
Over 80% of MED cases are immunologically mediated,3 including those clinically manifested even before the diagnosis of underlying pathologies. Some patients developed MED after beginning use of immunosuppressants, or with an increase in dose, suggesting that it was a reactive process rather than a simple benign neoplasia, and allowing for the establishment of a causal correlation between medication and the development of MED,8 which could explain the progressive increase of these lesions in the patient.
Although its pathogenesis remains unknown, recent evidence demonstrates the existence of several fibroblast growth factors derived from mast cells in patients with SLE and MED. 9
Yamamoto has demonstrated an increased numbers of mast cells both in the solitary dermatofibroma and in the MED. Mast cells are rich in cytokines that may affect fibroblasts, keratinocytes or t-cells, and may possibly induce several pathological changes, including epidermal acanthosis, basal melanosis, and the onset of the fibrosis process. In solitary dermatofibromas and in those with spontaneous regression, the number of those cells is lower than that in MED. 9 Immunohistochemistry shows positivity for antibodies against factor XIIIa, vimentin and actin. The transforming growth factor-beta (TGF-beta) can be a trigger for fibrosis. 10
The authors have described a case of a patient with MED, which began 20 years before the onset of SEL. No previous description of such a long period between the diagnosis of MED and that of an underlying disease could be found in the literature review carried out by the authors (Chart 1), which is presented in this paper.
1. Pinto-Almeida T, Caetano M, Alves R, Selores M. Congenital multiple clustered dermatofibroma and multiple eruptive dermatofibromas-unusual presentations of a common entity. An Bras Dermatol. 2013;88(6 Suppl 1):S63-6.
2. Yamamoto T. Katayama I, Nishioka K. Role of mast cells in dermatofibroma: recent viewpoints into the pathogenesis. Eur J Dermatol. 2003;13(5):419-23.
3. Taborda ML, Buffon RB, Bonamigo RR. Dermatofibromas eruptivos múltiplos e infecção pelo HCV tratada com Interferon. Revista da AMRIGS. 2006;50(4):334-6.
4. Niiyama S, Katsuoka K, Happle R, Hoffmann R. Multipleeruptive dermatofibromas: a review of the literature. Acta Dermato Venereol.2002;82(4):241-4.
5. Newman DM, Walter JB. Multiple dermatobromas inpatients with systemic lupus erythematosus on immunosuppressive therapy. N Engl J Med. 1973;289(160:842-3.
6. Bittencourt MJS, Miranda MFR, Parijós AM, Mesquita LB, Dermatofibroma sob pigmento preto de tatuagem: relato de um caso. An Bras Dermatol. 2013;88(4):625-7.
7. Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria forsystemic lupus erythematosus. Arthritis Rheum. 2012;;64(8):2677-86.
8. García-Millán C1, Aldanondo I, Fernández-Lorente M, Carrillo R, Jaén P. Multiple Eruptive Dermatofibromas in 2 Patients Infected With the Human Immunodeficiency Virus. Actas Dermosifiliogr. 2007;98(10):702-6.
9. Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literaure. Int J Dermatol.2008;47(7):723¬7.
10. Gencoglan G, Karaarslan IK, Dereli T, Kazandi AC. Dermatofibroma on the palmar surface of the hand. Skinmed.2008;7(1):41-3.
The present study was carried out at the Universidade Federal do Vale do São Francisco - Petrolina (PE), Brazil.
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}