


Tânia Rita Moreno de Oliveira Fernandes1; Graziele Áquila de Souza Brandão2; Talita Suzany Siqueira dos Santos3
Keywords: HISTIOCITOMA FIBROSO BENIGNO; LÚPUS ERITEMATOSO SISTÊMICO; IMUNOSSUPRESSORES.
Dermatofibromas solitários ou histiocitomas fibrosos benignos superficiais são tumorações fibro-histiocitárias dérmicas benignas e comuns que se apresentam como pápulas ou nódulos de coloração eritêmato-acastanhada, únicas ou em pequeno número, e em geral assintomáticas. O diagnóstico é clínico e histopatológico, e o tratamento é cirúrgico, recomendado apenas por razões estéticas e se houver sintomas, o que é incomum.1 A etiologia é desconhecida, a localização tem predileção pelos membros inferiores, e não há associação com doenças sistêmicas.1
Dermatofibromas eruptivos múltiplos (DEM) constituem uma variante que ocorre em 0,3% dos pacientes, com presença de 15 ou mais lesões, ou surgimento de cinco a oito tumores em período inferior a quatro meses.1 As lesões são geralmente dolorosas e generalizadas, e têm sido associadas com doenças autoimunes, drogas imunossupressoras e alterações hormonais.2 Muitos desses pacientes apresentam alteração de seu estado imunológico, mais comumente Aids e lúpus eritematoso sistêmico (LES);3,4 entretanto, outras desordens imunológicas (dermatomiosite, síndrome de Sjögren, hepatite C) ou mieloprolifarerativas (linfomas cutâneos de células T) podem estar associadas aos DEM.4 Além disso, agentes antirretrovirais, drogas imunobiológicas (efalizumab) e fator de necrose antitumoral alfa também podem estar envolvidos. Portanto, o surgimento desse tipo de entidade deve suscitar a investigação de doença subjacente. Na literatura científica foram descritos vários subtipos histológicos de dermatofibromas, mas em geral os DEM se apresentam histologicamente, como lesões pobremente circunscritas, exibindo hiperplasia da epiderme, proeminentes feixes de colágeno e difusa proliferação de fibrócitos.5Tratase de relato de caso que desenvolveu DEM 20 anos antes do diagnóstico de LES.
Paciente do sexo feminino, de 47 anos, parda, natural e procedente de Juazeiro (BA), relata o surgimento de aproximadamente cinco lesões nódulo-eritêmato-acastanhadas em membros inferiores (MMII), há 25 anos, negando histórico de sintomas locais ou sistêmicos e uso de medicamentos. Seguiu-se o surgimento gradual de outras lesões com igual característica, em MMII, membros superiores (MMSS) e abdômen, que continuam a aparecer até o presente momento.
Há cinco anos, apresentando febre de curso prolongado por aproximadamente 60 dias, poliartralgia, edema em articulações de mãos e pés, mialgias, inapetência, dor retroesternal e alopecia difusa, procurou o Serviço de Reumatologia.
Os exames laboratoriais revelaram: FAN 1: 1500 (padrão nuclear pontilhado fino); hemograma: Hb 10.7, leucócitos 8.000/ml, plaquetas 435.000; alfa-1 glicoproteína ácida 241 (N = até 117); alfa-1 antitripsina 282 (N = até 174); antiDNA nativo 1:10 (N = não reagente); VHS 52mm (na primeira hora); ecocardiograma: derrame pericárdico pequeno, com cavidades cardíacas e função diastólica normais; funções hepática e renal normais; PPD negativo.
Concretizado o diagnóstico de LES, a paciente foi medicada com meloxicam 15mg/dia, famotidina 30mg/dia, difosfato de cloroquina 200mg/dia, prednisona 20mg/dia e metotrexato 10mg/semana, evoluindo com desaparecimento da febre e melhora progressiva dos sintomas.
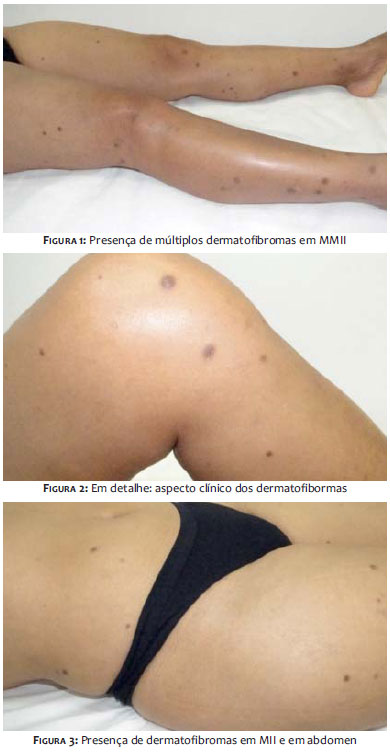
Dois anos após o surgimento do quadro sistêmico, procurou o Serviço de Dermatologia, por apresentar aumento significativo dos nódulos acastanhados, agora em número superior a 40, com predomínio em MMII, MMSS e abdômen (Figuras 1 a 3). A biópsia de uma das lesões, caracterizou dermatofibroma. (Figura 4).
Apresentava na ocasião FAN 1:1280, eletroforese de proteínas e ecocardiograma normais, encontrando-se em uso de meloxicam, famotidina e deflazacorte 6mg/dia.
Mantem-se, até o presente momento, acompanhada pelas clínicas de Reumatologia e Dermatologia, apresentando diminuição do FAN (1:320) e confirmando assim o controle do comprometimento sistêmico. Apresenta, no entanto, aumento progressivo dos DEM. Tendo sido esclarecida em relação à benignidade dos tumores, optou por não os retirar.
DEM foram associados a diversas comorbidades (56%)4 e doenças autoimunes tratadas com imunossupressores, especialmente LES (46%), infecção pelo HIV, colite ulcerativa, pênfigo vulgar, leucemia mieloide aguda e transplantes de órgãos, casos familiares e congênitos.5 Ocorre mais em pacientes do sexo feminino, o que talvez se explique pela ocorrência de doenças autoimunes, em especial LES, predominantemente em mulheres.
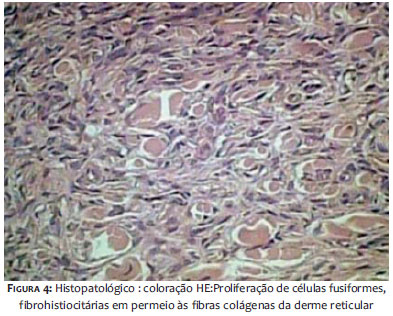
Histologicamente caracteriza-se por proliferação na derme de células fusiformes não encapsuladas com a periferia composta de feixes de colágeno. A epiderme sobrejacente apresenta-se acantótica e com hiperpigmentação da camada basal. É comum a presença de infiltrado inflamatório linfocitário.6
Nesse caso, a paciente foi diagnosticada com o DEM 20 anos após seu surgimento e, nesse intercurso, foi diagnosticada como LES pela presença de alopécia não cicatricial, artrite de duas ou mais articulações periféricas, serosite, FAN e antiDNA positivos, preenchendo, portanto, cinco dos critérios propostos pelo Systemic Lupus International Collaborating Clinics que, em 2012, definiu novos critérios de classificação para LES, fundamentando o diagnóstico na presença de quatro dos 17 relacionados, sendo pelo menos um critério clínico e um imunológico, ou biópsia renal compatível com nefrite lúpica associada a FAN ou antiDNA positivos.7
Mais de 80% dos casos de DFM são mediados imunologicamente,3 inclusive manifestam-se clinicamente antes mesmo do diagnóstico de patologias subjacentes. Alguns pacientes desenvolveram DFM após início do uso de imunossupressor, ou aumento de sua dose, o que sugere tratar-se de processo reativo e não simples neoplasia benigna, e permite estabelecer uma relação causal entre medicação e o desenvolvimento dos DEM,8 o que pode justificar o aumento progressivo dessas lesões nessa paciente.
Apesar de sua patogênese permanecer desconhecida, evidências recentes demonstram a existência de vários fatores de crescimento de fibroblastos derivados de mastócitos em pacientes com LES e DEM.9
Yamamoto demonstrou aumento do número de mastócitos tanto no dermatofibroma solitário quanto no DEM. Mastócitos são células ricas em citocinas que podem afetar fibroblastos, queratinócitos ou células T e, possivelmente, induzir várias alterações histopatológicas, incluindo acantose da epiderme, melanose basal e o desencadeamento do processo de fibrose. Nos dermatofibromas solitários e naqueles em regressão espontânea, o número dessas células é inferior ao das referentes ao DEM.9 A imuno-histoquimica mostra positividade para anticorpos contra fator XIIIa, vimentina e actina. O fator transformador de crescimento beta (TGF-beta) pode ser um gatilho da fibrose.10
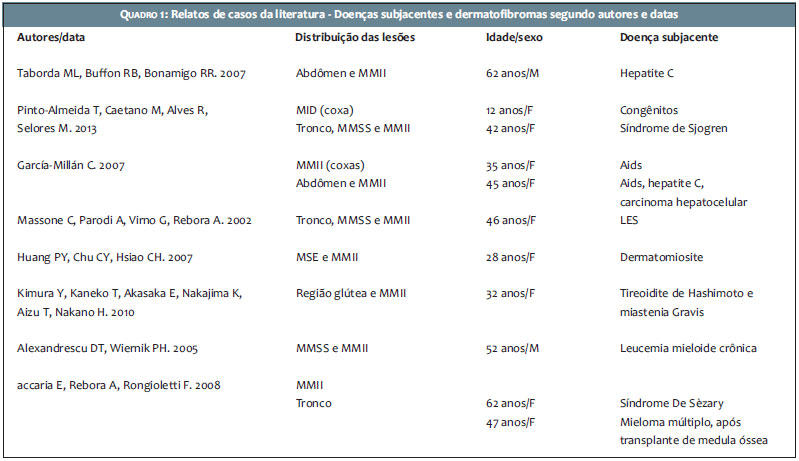
Relatamos o caso de paciente com DEM, iniciado 20 anos antes do surgimento de LES; não encontramos na literatura, da qual apresentamos uma síntese de revisão (Quadro 1), casos precedendo em tantos anos o diagnóstico de alguma doença de base.
1. Pinto-Almeida T, Caetano M, Alves R, Selores M. Congenital multiple clustered dermatofibroma and multiple eruptive dermatofibromas-unusual presentations of a common entity. An Bras Dermatol. 2013;88(6 Suppl 1):S63-6.
2. Yamamoto T. Katayama I, Nishioka K. Role of mast cells in dermatofibroma: recent viewpoints into the pathogenesis. Eur J Dermatol. 2003;13(5):419-23.
3. Taborda ML, Buffon RB, Bonamigo RR. Dermatofibromas eruptivos múltiplos e infecção pelo HCV tratada com Interferon. Revista da AMRIGS. 2006;50(4):334-6.
4. Niiyama S, Katsuoka K, Happle R, Hoffmann R. Multipleeruptive dermatofibromas: a review of the literature. Acta Dermato Venereol.2002;82(4):241-4.
5. Newman DM, Walter JB. Multiple dermatobromas inpatients with systemic lupus erythematosus on immunosuppressive therapy. N Engl J Med. 1973;289(160:842-3.
6. Bittencourt MJS, Miranda MFR, Parijós AM, Mesquita LB, Dermatofibroma sob pigmento preto de tatuagem: relato de um caso. An Bras Dermatol. 2013;88(4):625-7.
7. Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria forsystemic lupus erythematosus. Arthritis Rheum. 2012;;64(8):2677-86.
8. García-Millán C1, Aldanondo I, Fernández-Lorente M, Carrillo R, Jaén P. Multiple Eruptive Dermatofibromas in 2 Patients Infected With the Human Immunodeficiency Virus. Actas Dermosifiliogr. 2007;98(10):702-6.
9. Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literaure. Int J Dermatol.2008;47(7):723¬7.
10. Gencoglan G, Karaarslan IK, Dereli T, Kazandi AC. Dermatofibroma on the palmar surface of the hand. Skinmed.2008;7(1):41-3.
Trabalho realizado na Universidade Federal do Vale do São Francisco - Petrolina (PE), Brasil.
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}