


Melissa de Almeida Corrêa Alfredo1; Priscila Neri Lacerda1; Ana Flávia Teixeira Abreu1; Alexandre Morais Carneiro2; Eloisa Bueno Pires Campos1; Helio Amante Miot1
Financial support: None
Conflict of interest: None
How to cite this article: Alfredo MAC, Lacerda PN, Abreu AFT, Carneiro AM, Campos EBP, Miot HA. Extraskeletal Ewing sarcoma: report of a rare and exuberant case simulating lipoma. Surg Cosmet Dermatol. 2023;15:e20230183.
Extraskeletal Ewing sarcoma (EES) is a rare tumor that affects men between 10-20 years old and represents 25% of Ewing sarcomas. We report a case due to the previous diagnosis of a fast-growing lipoma to highlight the importance of histopathological diagnosis. A 14-year-old boy presented 10x10 cm tumor in the right shoulder for a year with ultrasonography suggesting lipoma. The lesion was excised and EES was confirmed by immunohistochemical analysis. EES diagnosis in young people is challenging compared to subcutaneous tumors, and histopathology is essential. The rapid progression of the tumor and high metastatic rates highlight the significance of early treatment.
Keywords: Sarcoma, Ewing; Neuroectodermal tumors; Sarcoma
Ewing sarcoma is the second most common malignant bone neoplasm in the pediatric population after osteosarcoma. It occurs mainly in adolescents in the second decade of life and has high metastasis rates.1 James Ewing described it in 1921, and it constitutes 10-15% of all bone sarcomas, including classic Ewing sarcoma, extraskeletal Ewing sarcoma (EES), malignant small cell tumor of the chest wall, and primitive neuroectodermal tumors of soft tissues (PNET), all with differentiation from mesenchymal stem cells.2 Its histological origin is not well elucidated, and, to date, there are no well-established associations with family history, environmental exposure, or radiation history.1,2 Genetically, they are characterized by spontaneous chromosomal translocations, and the most specific for EES are t(11;22)(q24;q12) and t(21;22)( q24;q12).1,2,3
EES presents as a rare, primary soft tissue tumor, accounting for approximately 25% of the reported cases of Ewing Sarcoma.3 It has a poor prognosis depending on the stage, and it is prevalent in men in the second and third decades of life.4 We report the case of EES in a 14-year-old patient with a previous diagnosis of rapidly evolving lipoma to highlight the importance of early investigation, differential diagnoses, and histopathological study to achieve curative therapeutic possibilities and a better prognosis.
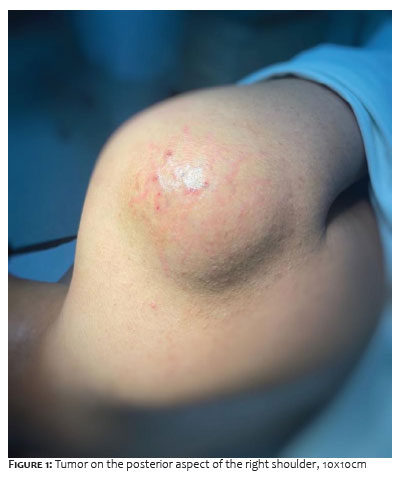
A 14-year-old man, eutrophic and without comorbidities, was referred to the tertiary service of Dermatology due to the appearance of a tumor in the right shoulder one year ago, with progressive growth, presenting pain, and limitation of movement. Previous ultrasonography showed a hypoechoic nodular formation measuring 4.5 x 1.4 cm, intermingled with fine fibrous strands, without significant vascular flow, located in the subcutaneous region of the right shoulder, with characteristics suggestive of lipoma. Due to the Covid-19 pandemic, the patient was seen nine months after the imaging exam, showing exuberant enlargement of the tumor, measuring 10 x 10 cm in the posterior region of the right shoulder (Figure 1). Immediate exeresis of the lesion was performed, which, during the intraoperative period, appeared as a violaceous tumor, somewhat friable and adhered to deep planes (Figure 2).
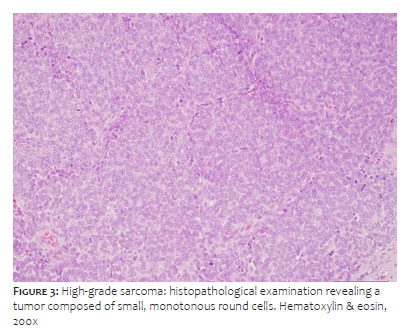
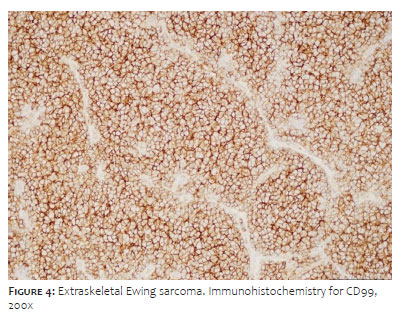
Histopathological analysis revealed a multinodular growth pattern composed of small, monotonous, round to ovoid cells, containing scarce cytoplasm and hyperchromatic nuclei organized in nests delimited by thin fibrous septa. We observed a markedly hypercellular tumor with an exuberant pseudoangiomatous pattern. Abundant mitotic figures were identified, in addition to multiple foci of necrosis (Figure 3). The immunohistochemical study showed immunoexpression for CD99 and FLI1, focal expression of cytokeratin (AE1/AE3) and S100, in addition to negativity for CD34, smooth muscle actin, desmin, myophenin, synaptophysin, EMA, and TLE1 (Figure 4). The set of findings confirmed the diagnosis of EES.
Oncological staging revealed bone marrow metastasis. We conducted extensive surgical expansion and chemotherapy with the stability of the condition. The patient continues with 12 months of follow-up in the 13th cycle of chemotherapy and the absence of other neoplastic foci/metastases.
EES is a rare malignant neoplasm that presents as a painful, fast-growing nodule, frequently located in the paravertebral regions and lower limbs.5 Histologically, it presents compact, homogeneous, small, round to oval cells arranged in sheets, showing in immunohistochemistry marked expression of CD99 on its surface.2,5 It tends to local dissemination and high rates of metastasis, with prognosis dependent on tumor extension and the presence of metastases at diagnosis.6 Thus, early diagnosis through histopathology becomes essential for the aggressive approach to the tumor and improvement of survival.6
Appropriate treatment involves surgery and/or local radiotherapy associated with chemotherapy.6,7 Although EES is considered radiosensitive currently, wide surgical excision is the local therapeutic method of choice due to the adverse events associated with radiotherapy.6,7 The complete resection of the tumor associated with early age at diagnosis is considered a predictor of better prognosis and longer survival.6,7 On the other hand, age greater than 14 years at diagnosis, primary tumor volume greater than 200 mL, and metastases (mainly in the bone marrow and lung) are the main factors associated with a worse prognosis.6,7 Chemotherapy with anthracyclines can be used as adjuvant therapy, and the role of local adjuvant radiotherapy after complete resection is still inconclusive, although it has been shown to improve survival. Neoadjuvant chemotherapy seems to achieve promising results, but it depends on prospective clinical trials.8
The diagnosis of EES in a young patient is challenging, both because of its rarity and the plethora of diseases that can manifest as subcutaneous nodules. Differential diagnoses in adolescence make the histopathological study essential, and the rapid progression of the tumor associated with high metastatic rates highlights the importance of early multimodal therapy. Despite marked improvements in survival, a better understanding of the complex biology of extraskeletal Ewing sarcoma may provide a roadmap for the successful development of biologically targeted therapies.
Melissa de Almeida Corrêa Alfredo 0000-0001-7423-4190
Approval of the final version of the manuscript; preparation and writing of the manuscript; data collection, analysis, and interpretation; intellectual participation in propaedeutic and/or therapeutic conduct of studied cases; critical literature review.
Priscila Neri Lacerda 0000-0001-8100-5978
Preparation and writing of the manuscript; intellectual participation in propaedeutic and/or therapeutic conduct of studied cases; critical literature review.
Ana Flávia Teixeira Abreu 0000-0003-4169-9068
Preparation and writing of the manuscript; data collection, analysis, and interpretation; intellectual participation in propaedeutic and/or therapeutic conduct of studied cases; critical literature review.
Alexandre Morais Carneiro 0000-0002-8947-8611
Data collection, analysis, and interpretation; intellectual participation in propaedeutic and/or therapeutic conduct of studied cases.
Eloisa Bueno Pires Campos 0000-0001-7185-4650
Data collection, analysis, and interpretation; intellectual participation in propaedeutic and/or therapeutic conduct of studied cases.
Helio Amante Miot 0000-0002-2596-9294
Approval of the final version of the manuscript; study design and planning; active participation in research orientation; critical literature review; critical revision of the manuscript.
1. Durer S, Shaikh H. Ewing Sarcoma. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022.
2. Ludwig JA. Ewing sarcoma: historical perspectives, current state-of-the-art, and opportunities for targeted therapy in the future. Curr Opin Oncol. 2008;20(4):412–8.
3. Abboud A, Masrouha K, Saliba M, Haidar R, Saab R, Khoury N, et al. Extraskeletal Ewing sarcoma: diagnosis, management and prognosis. Oncol Lett. 2021;21(5):354.
4. Eloqayli H. Adult primary cervical extra-osseous Ewing's sarcoma: a case report and short literature review. Int J Surg Case Rep. 2017;41:83-5.
5. Kennedy JG, Eustace S, Caulfield R, Fennelly DJ, Hurson B, O'Rourke KS. Extraskeletal Ewing's sarcoma: a case report and review of the literature. Spine (Phila Pa 1976). 2000;25(15):1996-9.
6. Ladenstein R, Pötschger U, Le Deley MC, Whelan J, Paulussen M, Oberlin O, et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol. 2010;28(20):3284-91.
7. Dunst J, Schuck A. Role of radiotherapy in Ewing tumors. Pediatr Blood Cancer. 2004;42(5):465-70.
8. Dirksen U, Ranft A, Baumhoer D, Berg H van den, Brichard B, Eich H-T, et al. Association of treatment delays with an unfavorable outcome in patients with localized Ewing sarcoma: a retrospective analysis of data from the GPOH Euro - E.W.I.N.G. 99 trial. 2021;39:11502.
 All content the journal, except where identified, is under a Creative Commons Attribution-NonCommercial 4.0 International license - ISSN-e 1984-8773
All content the journal, except where identified, is under a Creative Commons Attribution-NonCommercial 4.0 International license - ISSN-e 1984-8773



 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}