


Helio Amante Miot; Cesar Augusto Zago Ferreira; Ana Claudia Athanasio Shwetz; Luciane Donida Miot; Gabriela Roncada Haddad
Fonte de financiamento: Nenhuma
Conflito de interesses: Nenhum
Como citar este artigo: Miot HA, Ferreira CAZ, Shwetz ACA, Miot LD, Haddad GR. Histiocitoma fibroso maligno em tornozelo: relato de caso. Surg Cosmet Dermatol. 2021;13:e20210028
Histiocitoma fibroso maligno (MFH) ou sarcoma pleomórfico indiferenciado (UPS) é um sarcoma moderadamente agressivo, capaz de invadir estruturas adjacentes. Trata-se de neoplasia mesenquimal que predomina em homens entre a sexta e sétima décadas de vida. Localiza-se, principalmente, nos membros inferiores, podendo acometer cabeça e pescoço, tronco e retroperitônio, com tendência à recorrência e à metástase local. O presente relato tem como objetivo apresentar um caso de MFH no tornozelo de uma mulher de 49 anos, com invasão óssea adjacente, que evoluiu com amputação transtibial. São abordados aspectos clínicos, radiológicos, histopatológicos e terapêuticos, salientando-se a importância do diagnóstico precoce.
Keywords: Amputação; Histiocitoma fibroso maligno; Sarcoma
Histiocitoma fibroso maligno (MFH) foi descrito por O´Brian e Staut em 19641, caracterizando-se por neoplasia mesenquimal de alto grau, composta por fibroblastos, miofibroblastos e histiócitos.2 Predomina entre a sexta e sétima décadas de vida e em homens (2/3 dos casos).3,4
Acomete, principalmente, os membros inferiores, sendo o fêmur distal, fíbula proximal e fêmur proximal2 as topografias mais frequentes, apesar de ocorrer também em: pulmão, rim, bexiga, vias deferentes, coração, aorta, estômago, intestino delgado, órbita, sistema nervoso central, medula espinhal, seios da face, cavidades nasais e oral.5
O MFH é associado a doenças hematopoiéticas (linfoma não Hodgkin, linfoma de Hodgkin, mieloma múltiplo e histiocitose maligna) ou decorrente de radiação, fratura, osteonecrose, doença de Paget, fibroma não ossificante ou displasia fibrosa, sendo, nesses casos, mais agressivo.5
Apresenta comportamento moderadamente agressivo, capaz de invadir tecidos moles adjacentes, sistema esquelético e retroperitônio. O principal fator prognóstico é o estágio clínico do tumor, definido pelo grau de diferenciação, tamanho e presença de metástases à distância. Ainda, subtipo histológico, local anatômico, profundidade do tumor e tratamento realizado também influenciam o prognóstico.6
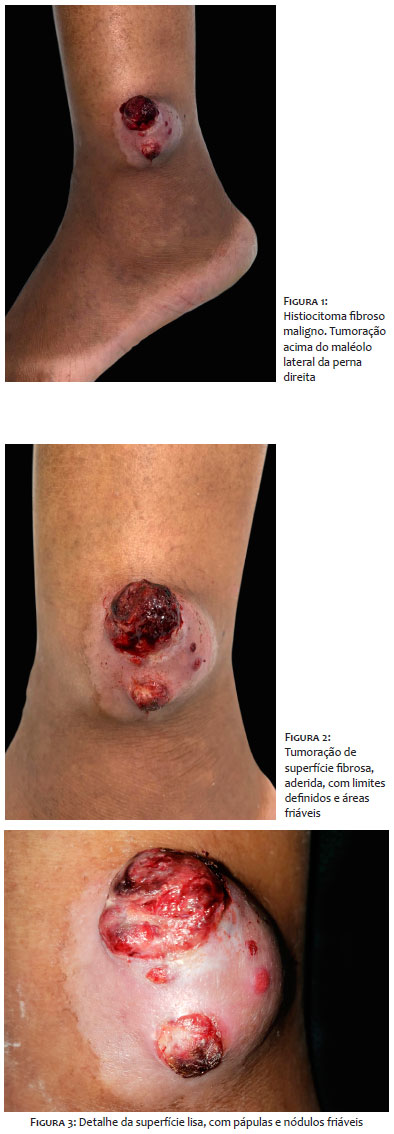
Mulher, 49 anos, negra, dona de casa, apresentava tumoração friável, com superfície sangrante, consistência fibrosa, aderida, de contorno definido, localizada no maléolo lateral direito, medindo 7,0x5,5cm, de crescimento progressivo nos últimos nove meses (Figuras 1 a 3). Referia episódios de sangramento associados à dor local. Procurou assistência médica, sendo tratada como infecção bacteriana, com antibioticoterapia, sem melhora.
Apresentava como antecedentes: trombose de veia porta, insuficiência cardíaca e hipertensão arterial. Não foram identificados linfadenopatias, visceromegalias ou linfedema. Os diagnósticos aventados foram melanoma amelanótico, sarcoma e carcinoma de Merkel.
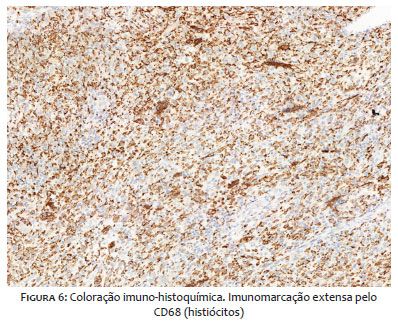
O exame histopatológico revelou neoplasia mesenquimal ulcerada, composta de células fusiformes e epitelioides, com pleomorfismo nuclear e atividade mitótica acentuada (Figuras 4 e 5), caracterizando MFH, de alto grau histológico. O painel imuno-histoquímico do tumor mostrou positividade para CD68 (Figura 6) e negatividade para p63, AE1/E3, CK5/6, MELAN A, CD34, AML e S100, SOX-10.
Os exames bioquímicos gerais foram normais, porém, à ressonância nuclear magnética, foi evidenciada a invasão da fíbula distal (Figura 7).
O conjunto de achados histopatológicos, imunofenotípicos e de imagem são diagnósticos de histiocitoma fibroso maligno (profundo)/sarcoma pleomórfico indiferenciado, estágio IIIA.
Em decorrência do acometimento ósseo e da irressecabilidade do tumor com preservação de estruturas profundas, optou-se pela amputação transtibial. A paciente se mantém em seguimento com a Oncologia há 11 meses, sem recidiva. Em decorrência da cardiopatia, foi contraindicada a quimioterapia adjuvante.
Sarcomas de tecidos moles são neoplasias raras, contabilizando aproximadamente 1% dos cânceres sólidos em adultos, que representam um heterogêneo grupo de desordens decorrentes de tecido mesenquimal.7
Em 2003, Coindre descreveu uma nova classificação para sarcomas, baseado em critérios imuno-histoquímicos:8
- Sarcomas com imuno-histoquímica definitiva: rabdomiossarcomas, sarcoma epitelioide, sarcoma de células claras, tumor desmoplásico de pequenas células redondas e tumores estromais gastrointestinais.
- Sarcomas com perfil imuno-histoquímico não específico: sarcoma de Ewing, leiomiossarcoma, tumor maligno da bainha do nervo periférico, dermatofibrossarcoma protuberans e fibroblastoma de células gigantes, condrossarcoma mixoide extraesquelético, lipossarcomas e sarcoma alveolar de partes moles.
- Sarcomas com imuno-histoquímica não específica (não exibem marcadores específicos): sarcomas mal diferenciados, como fibrossarcoma, mixofibrossarcoma e histiocitoma fibroso maligno. Nesses casos, a imuno-histoquímica é útil para excluir outros tumores não mesenquimais.
O sarcoma pleomórfico indiferenciado (UPS) pode ser dividido entre os subtipos superficial e profundo, também chamados fibroxantoma atípico (AFX) e histiocitoma fibroso maligno (MFH), respectivamente. A distinção entre as entidades é fundamental para predizer a agressividade locorregional do tumor e o seu prognóstico.9,10
Neste relato, o MFH ocorreu no tornozelo de uma mulher, adulta, entretanto esses tumores predominam em homens, maiores de 50 anos.4 Tipicamente, apresenta-se como tumoração indolor, de crescimento rápido, havendo relatos de lesões unicamente subcutâneas.11
O diagnóstico do UPS requer a diferenciação histopatológica de tumores, como: melanoma, carcinoma de células escamosas, angiossarcoma, leiomiossarcoma e outras neoplasias indiferenciadas.12,13
O MFH é reconhecido como um grupo heterogêneo de tumores que compartilham um fenótipo comum, sendo necessários imuno-histoquímica, microscopia eletrônica ou estudos moleculares para melhor caracterização.14
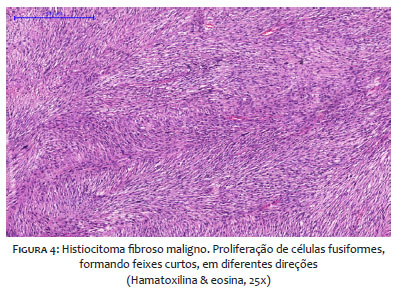
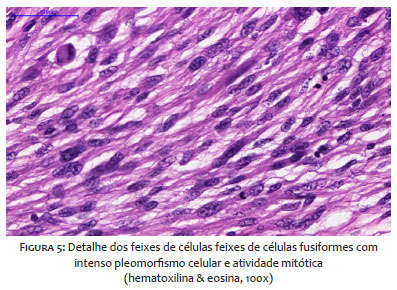
A histopatologia revela células fusiformes pleomórficas dispostas em feixes comuns, padrão estoriforme, podendo ser encontrados histiócitos multinucleados, infiltrados em derme profunda e tecido celular subcutâneo.15,16
O MFH apresenta imunorreatividade para vimentina e CD68 (marcador histiocítico).17,18 S100, desmina, S-100 e HMB-45 são anticorpos encontrados no lipossarcoma ou nervo periférico maligno, tumor de bainha, rabdomiossarcoma e melanoma maligno, respectivamente, ao passo que CD34 mostra reatividade em angiossarcomas.15,16
Ressonância magnética é a modalidade de imagem de escolha para avaliação de sarcomas de tecidos moles, especialmente para determinar a extensão local da lesão. Ao exame, o MFH mostra um padrão heterogêneo hiperdenso em imagens ponderadas em T2 e isodenso aos músculos em imagens ponderadas em T1.19
A excisão ampla (margem ≥2cm) é a abordagem recomendada. Contudo, dependendo da topografia, há limitação pela proximidade com estruturas nobres14, havendo indicação de cirurgia micrográfica.6,20
Altas taxas de recorrência local são relatadas em pacientes submetidos à excisão cirúrgica (19 a 66%).20-22 Neste caso, a lesão foi totalmente removida e, devido a comorbidades, optou-se por não realizar quimioterapia adjuvante.
Um estudo realizado com 167 pacientes identificou entre pacientes tratados com ressecção marginal que 66% apresentaram recorrência local, contrapondo-se a 27% dos submetidos à ressecção ampla (margem ≥3cm).23
Radioterapia pode ser útil como um tratamento adjuvante, sendo empregado um campo de radiação abrangendo o local do tumor e 5cm ao redor, com doses que variam entre 50 e 65 Gray. Entretanto, o impacto geral na recorrência e sobrevida não é completamente definido.24
Quimioterapia é tipicamente empregada para doenças generalizadas, porém com prognósticos pouco expressivos.25,26 Atualmente, estudos com imunobiológicos em pacientes com doenças avançadas estão em fase clínica, como é o caso de ipilimumabe (anti-CTLA-4), em 33 crianças e adultos jovens, e sunitinibe (oral multi-kinase inhibitor) em 48 adultos jovens com sarcoma irressecável ou metastático.27
O MFH apresenta clínica indistinguível de outras entidades, sendo caracterizado principalmente pelo comportamento agressivo de crescimento. O presente relato de caso objetivou salientar suas principais características e a importância da suspeição diagnóstica pelo dermatologista e do tratamento precoce, a fim de se evitarem desfechos desfavoráveis.
Helio Amante Miot 0000-0002-2596-9294
Aprovação da versão final do manuscrito; revisão crítica da literatura; participação efetiva na orientação da pesquisa; participação intelectual no manejo propedêutico e/ou terapêutico dos casos estudados; revisão crítica do manuscrito; preparação e redação do manuscrito; concepção e planejamento do estudo.
Cesar Augusto Zago Ferreira 0000-0001-7299-1710
Aprovação da versão final do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito; preparação e redação do manuscrito; concepção e planejamento do estudo.
Ana Claudia Athanasio Shwetz 0000-0002-9373-5019
Contribuição do autor: Aprovação da versão final do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Luciane Donida Miot 0000-0002-2388-7842
Aprovação da versão final do manuscrito; revisão crítica da literatura; participação efetiva na orientação da pesquisa; participação intelectual no manejo propedêutico e/ou terapêutico dos casos estudados; revisão crítica do manuscrito.
Gabriela Roncada Haddad 0000-0002-7516-9586
Aprovação da versão final do manuscrito; revisão crítica da literatura; participação efetiva na orientação da pesquisa; participação intelectual no manejo propedêutico e/ou terapêutico dos casos estudados; revisão crítica do manuscrito.
1. O'Brien JE, Stout A P. Malignant fibrous xanthomas. Cancer. 1964;17:1445-55.
2. Pobirci DD, Bogdan F, Pobirci O, Petcu CA, Roşca E. Study of malignant fibrous histiocytoma: clinical, statistic and histopatological interrelation. Rom J Morphol Embryol. 2011;52(1 Suppl):385-8.
3. Marcet S. Atypical fibroxanthoma/malignant fibrous histiocytoma. Dermatol Ther. 2008;21(6):424-7.
4. Clark DW, Moore BA, Patel SR, Guadagnolo BA, Roberts DB, Sturgis EM. Malignant fibrous histiocytoma of the head andneck region. Head Neck. 2011;33(3):303-8.
5. Amjad M, Bari A. Malignant fibrous histiocytoma: an uncommon soft tissue sarcoma. J Pakistan Assoc Dermatol. 2009;19(4):243-6
6. Huether MJ, Zitelli JA, Brodland DG. Mohs micrographic surgery for the treatment of spindle cell tumors of the skin. J Am Acad Dermatol. 2001;44(4):656-9.
7. Clark MA, Fisher C, Judson I, Thomas JM. Soft-tissue sarcomas in adults. N Engl J Med. 2005;353(7):701-11.
8. Coindre JM. Immumo histochemistry in the diagnosis of soft tissue tumours. Histopathology. 2003;43(1):1-16.
9. Fletcher CD. The evolving classification of soft tissue tumours an update based on the new 2013 WHO classification. Histopathology. 2014;64(1):2-11.
10. Henderson MT, Hollmig ST. Malignant fibrous histiocytoma: changing perceptions and management challenges. J Am Acad Dermatol. 2012;67(6):1335-41.
11. Luzar B, Calonje E. Morphological and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37(3):301-9.
12. Pujani M, Hassan MJ, Jetley S. Atypical fibroxanthoma in a young female misdiagnosed clinically as a malignant melanomadan - An unusual presentation. J Cancer Res Ther. 2015;11(4):1027.
13. Deyrup AT, Haydon RC, Huo D, Ishikawa A, Peabody TD, He TC, et al. Myoid differentiation and prognosis in adult pleomorphic sarcomas of the 203 extremity: an analysis of 92 cases. Cancer. 2003;98(4):805-13.
14. Vassiliou A, Vlastarakos PV, Manolopoulos L, Yiotakis I, Voros D, Carvouni E, et al. Metastatic sarcoma of the tongue: pleomorphic malignant fibrous histiocytoma and literature review. J Rhinol Otol. 2014;2(1):10-3.
15. Winchester D, Lehman J, Tello T, Chimato N, Hocker T, Kim S, et al. Undifferentiated pleomorphic sarcoma: factors predictive of adverse outcomes. J Am Acad Dermatol. 2018;79(5):853-9.
16. Al-Agha OM, Igbokwe AA. Malignant fibrous histiocytoma: between the past and the present. Arch Pathol Lab Med. 2008;132(6):1030-5.
17. Dei Tos AP. Classification of pleomorphic sarcomas: where are we now? Histopathology. 2006;48(1):51-62.
18. Park SW, Kim HJ, Lee JH, Ko YH. Malignant fibrous histiocytoma of the head and neck: CT and MR imaging findings. AJNR Am J Neuroradiol. 2009;30(1):71-6.
19. Brown MD, Swanson NA. Treatment of malignant fibrous histiocytoma and atypical fibroxanthomas with micrographic surgery. J Dermatol Surg Oncol. 1989;15(12):1287-92.
20. Iorizzo LJ, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37(2):146-57.
21. Peiper M, Zurakowski D, Knoefel WT, Izbicki JR. Malignant fibrous histiocytoma of the extremities and trunk: an institutional review. Surgery. 2004;135(1):59-66.
22. Kearney MM, Soule EH, Ivins JC. Malignant fibrous histiocytoma: a retrospective study of 167 cases. Cancer. 1980;45(1):167-78.
23. Mundt AJ, Awan A, Sibley GS, Simon M, Rubin SJ, Samuels B, et al. Conservative surgery and adjuvant radiation therapy in the management of adult soft tissue sarcoma of the extremities: clinical and radiobiological results. Int J Radiat Oncol Biol Phys. 1995;32(4):977-85.
24. Marchese R, Bufo P, Carrieri G, Bove G. Malignant fibrous histiocytoma of the kidney treated with nephrectomy and adjuvant radiotherapy: a case report. Case Report Med. Epub 2010.
25. Tarjan M, Cserni G, Szabo Z. Malignant fibrous histiocytoma of the kidney. Scand J Urol Nephrol. 2001;35(6):518-20.
26. ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2011 Oct 3 - . Identifier NCT01445379, Phase I Study of Ipilimumab (Anti-CTLA-4) in Children and Adolescents With Treatment-Resistant Cancer. Available from: https://clinicaltrials.gov/ct2/show/NCT00556881
27. ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2006 Nov 17 - . Identifier NCT00400569, Phase II Study of Sunitinib Malate for Metastatic and/or Surgically Unresectable Soft Tissue Sarcoma. Available from: 240 https://clinicaltrials.gov/ct2/show/NCT00400569
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}