


Marcia Ferraz Nogueira; Jayme de Oliveira Filho; Gabriela Machado Dias Junqueira; Ana Julia Marçal Pereira Dias
Received on: 03/03/2020
Approved on: 02/03/2021
Financial support: None
Conflict of interest: None
Acknowledgments: We thank to Dr. Monica Freitas for her collaboration with the patient’s photos
Study conducted at the 1Department of Dermatology of the Faculty of Medicine of the Universidade de Santo Amaro, São Paulo (SP), Brazil
Neurofibromatosis type I (NF1) is an autosomal dominant disease, with an incidence of 1/2,500-3,000 births and a prevalence of approximately 1/4,000-5,000 individuals. Genetic mutations in the NF1 gene cause it, affecting neural and skin tissues. Glomus tumor is a benign neoplasm originating from the glomus, a neuromyoatrial structure of the skin present at the fingertips and involved in thermoregulation. The literature historically considered these tumors isolated and sporadic, but some studies have proved a relationship with neurofibromatosis type I. Thus, patients with neurofibromatosis type 1 should be investigated. The case report provides additional support for the notion that NF1 has a risk associated with multiple glomus tumors.
Keywords: Glomus Tumor; Association; Case Reports; Neoplasms; Neurofibromatoses
Glomus tumors are extremely painful benign tumors of the glomus body, a neuromyoarterial structure that exists in high concentrations in the fingertips, and is involved in thermoregulation. Glomus tumors are usually solitary and often arise in a subungual location, although multifocal disease and nonsubungual presentation are not uncommon. They occur more commonly in women and usually in the fourth decade of life.1 Although the true incidence of glomus tumors is unknown, they represent less than 2% of primary hand tumors.2
Historically, they have been considered isolated sporadic tumors, not associated with other disease processes. However, multiple case reports, a molecular genetics research, and an epidemiologic study have confirmed that neurofibromatosis type I is associated with glomus tumors.1
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder, with an incidence of 1/2,500–3,000 births and a prevalence of approximately 1/4,000-5,000 individuals.3,4 It is caused by mutations in the NF1 tumor suppressor gene, located on chromosome 17 (17q11.2), which encodes neurofibromin (nf), a protein able to downregulate the Ras-Raf/MAPK signaling pathway that activates cell proliferation. Mutations of the NF1 gene result in function alteration or loss of negative regulator of growth and cellular differentiation of nf. It leads to uncontrolled cell proliferation and an increased risk of developing cancer.4,5
A 26-year-old woman with NF1 was assisted at our institution complaining of severe pain in the subungual region of the fifth right finger, on the hyponychium and the lateral nail fold of the third left finger, and the subungual region of the left hallux.
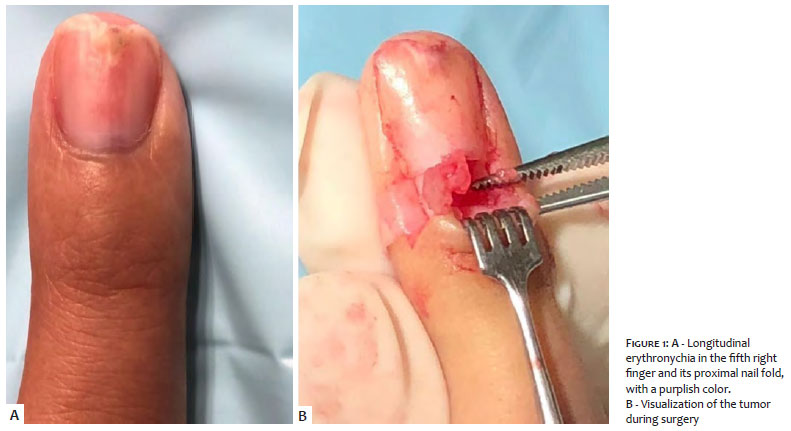
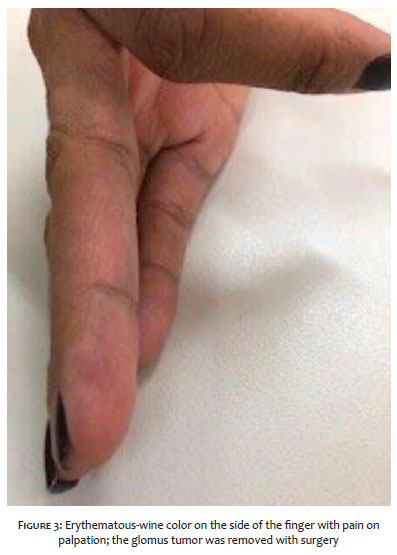
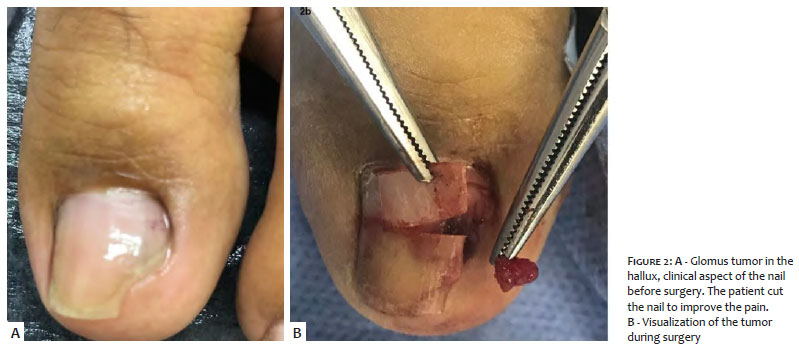
Clinically, the fifth right finger presented longitudinal erythronychia, with purple color and local pain in the proximal nail fold (Figures 1 and 3). The examination of the third left finger was normal, while in the left hallux, we noticed a purple tumor on the middle of the nail bed (Figure 2).
She presented the triad of Carroll (severe pain, point tenderness, and cold sensitivity) in all fingers.
A high-frequency ultrasound showed a 6 mm tumor on the fifth right subungual finger.
After troncular anesthesia, an oblique incision was made in the lateral portion on both sides of the proximal nail fold, exposing the nail matrix. We conducted the avulsion of the nail and incision in the nail bed, visualizing a reddish tumor, easily detached by divulsion using mosquito forceps. Histological examination of the tumor confirmed the diagnosis of glomus tumor.
Glomus tumors, usually present in the fingers and toes, are benign tumors that develop from cells that resemble the modified smooth muscle cells of the glomus apparatus.6 NF1, an autosomal dominant inherited disorder, is caused by mutations of the NF1 gene located on chromosome 17. The protein encoded by the NF1 gene, neurofibromin, acts as a tumor suppressor. Patients with NF1 lose neurofibromin expression, which leads to increased cell proliferation associated with protein kinase, with a propensity to develop many different types of tumors.7
With four glomus tumors excised, the present case indicates, together with some previously reported cases, that there is an association between glomus tumors and NF1.
In 1938, Klaber provided the first report of a glomus tumor arising in a patient with neurofibromatosis.8 The literature no longer contains reports of glomus tumors in patients with neurofibromatosis until 1995, when Sawada et al.3 reported three patients with neurofibromatosis type I and subungual glomus tumors. Between 1995 and 2013, another 13 case reports described glomus tumors arising in the setting of neurofibromatosis type I. Although many of the authors speculated about the possibility of an association, it was not confirmed until 2009,1 when Brems et al.9 firmly established a causal relationship between NF1 and glomus tumors. Glomus tumors associated with NF1 exhibit biallelic inactivation of the NF1 gene; thus, seven of the 12 tumors assessed harbored gene and somatic mutations, while two sporadic glomus tumors did not present abnormalities in the NF1 gene.
In 2013, Harrison et al.2 conducted an epidemiological case-control study comparing a cohort of patients undergoing excision of a glomus tumor with a similar cohort of patients who underwent excision of other benign hand injuries. The study found that 29% of patients undergoing excision of the glomus tumor had a diagnosis of neurofibromatosis, while none of the patients in the control cohort had the disorder.
Most glomus tumors are unique. It is improbable that there will be synchronous tumors in adjacent fingers, as in the present case. However, in a patient with NF1, the risk of glomus tumors is increased. As this case exemplifies, the diagnosis of multifocal glomus tumors should be considered in these patients, especially when the classic triad of clinical features (spontaneous paroxysmal pain, point tenderness, and hypersensitivity to cold) is present. Most are located in the subungual area. Delayed diagnosis and treatment can lead to unnecessary debilitation in such patients.10
The case report provides additional support for the concept that NF1 has a risk associated with multiple glomus tumors. For dermatologists who manage patients with NF1, knowing this association can facilitate early diagnosis and appropriate treatment.
Marcia Ferraz Nogueira | 0000-0001-7872-7304
Approval of the final version of the manuscript; study design and planning; preparation and writing of the manuscript; active participation in research orientation; intellectual participation in propaedeutic and/or therapeutic conduct of studied cases; critical literature review; critical revision of the manuscript.
Jayme de Oliveira Filho | 0000-0003-0239-0981
Approval of the final version of the manuscript; study design and planning; active participation in research orientation.
Gabriela Machado Dias Junqueira | 0000-0003-0899-9341
Study design and planning; preparation and writing of the manuscript; data collection, analysis, and interpretation; active participation in research orientation; intellectual participation in propaedeutic and/or therapeutic conduct of studied cases; critical literature review.
Ana Julia Marçal Pereira Dias | 0000-0002-7063-3298
Study design and planning; preparation and writing of the manuscript; intellectual participation in propaedeutic and/or therapeutic conduct of studied cases; critical literature review.
1. Harrison B. Tumores Sammer D. Glomus e Neurofibromatose: uma associação recentemente reconhecida. Open Glob Cir Plást Reconstr. 2014;2(9):e214.
2. Harrison B, Moore AM, Calfee R, et al. A associação entre tumores glômicos e neurofibromatose. J Hand Surg Am. 2013;38:1571-4.
3. Huson S. Neurofibromatose: fenótipos emergentes, mecanismos e a gestão. Clin Med. 2008;8(6):611.
4. Scaravilli G, Rossi R, Artiaco S, Merolla G. Glomus tumor da eminência tenar na neurofibromatose tipo 1: relato de caso e revisão de literatura. Med Transl UniSa . 2015;11:63-8.
5. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Diretrizes para o diagnóstico e tratamento de indivíduos com neurofibromatose 1. J Med Genet. 2007;44(2):81-8.
6. Weedon D. Glomus tumor. In: Weedon D, editor. Patologia da pele de Weedon, 3th ed. China: Churchill Livingstone Elsevier; 2010. p. 907-8.
7. Brems H, Beert E, Ravel T, Legius E. Mecanismos na patogênese de tumores malignos na neurofibromatose tipo 1. Lancet Oncol. 2009;10:508-15.
8. Klaber R. Morbus recklinghausen com tumores glomóides. Proc R Soc Med. 1938; 31:347.
9. Brems H, Park C, Maertens O, Pemov A, Messiaen L, Upadhyaya M, et al. Tumores glômicos na neurofibromatose tipo 1: evidência genética, funcional e clínica de uma nova associação. Cancer Res 2009;69:7393-401.
10. Aqil N, Gallouj S, Moustaide K, Mernissi F. Tumores dolorosos em um paciente com neurofibromatose tipo 1: relato de caso. J Med Case Rep. 2018;12:319.
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}