


Marcia Ferraz Nogueira; Jayme de Oliveira Filho; Gabriela Machado Dias Junqueira; Ana Julia Marçal Pereira Dias
Data de recebimento: 03/03/2020
Data de aprovação: 02/03/2021
Suporte Financeiro: Nenhum
Conflito de Interesses: Nenhum. Agradecimentos: À Dra. Monica Freitas por sua colaboração com as fotos da paciente
Trabalho realizado no Departamento de Dermatologia da Faculdade de Medicina de Santo Amaro, São Paulo (SP), Brasil
Neurofibromatose tipo I (NF1) é uma doença autossômica dominante, com incidência de 1/2.500-3.000 nascimentos e prevalência de aproximadamente 1/4.000-5.000 indivíduos; é causada por mutações genéticas no gene NF1, que afetam tecidos neurais e cutâneos. Tumor glômico é uma neoplasia benigna originada do glomo, uma estrutura neuromioatrial da pele presente nas pontas dos dedos e envolvida na termorregulação. São considerados historicamente tumores isolados esporádicos, porém existem estudos que comprovam sua relação com a neurofibromatose tipo I. Pacientes com neurofibromatose tipo 1 devem ser investigados. O relato de caso fornece suporte adicional à noção de que NF1 tem um risco associado a múltiplos tumores glômicos.
Keywords: Tumor Glômico; Associação; Relatos de Casos; Neoplasias; Neurofibromatoses
Os tumores glômicos são tumores benignos, extraordinariamente dolorosos, do corpo glômico, uma estrutura neuromioarterial que existe em altas concentrações na ponta dos dedos e está envolvida na termorregulação. Os tumores glômicos são geralmente solitários e surgem em uma localização subungueal, embora a doença multifocal e apresentação não subungueal não sejam incomuns. Ocorrem mais comumente em mulheres e, geralmente, na quarta década de vida.1 Embora a verdadeira incidência de tumores glômicos seja desconhecida, eles representam menos de 2% dos tumores primários da mão.2
Historicamente, eles são considerados tumores isolados esporádicos, não associados a outros processos patológicos. No entanto, relatos de casos múltiplos, um estudo de genética molecular e um estudo epidemiológico confirmaram que a neurofibromatose tipo I está associada a tumores glômicos.1
A neurofibromatose tipo 1 (NF1) é um distúrbio autossômico dominante, com incidência de 1/2.500–3.000 nascimentos e prevalência de aproximadamente 1/4.000-5.000 indivíduos.3,4 É causada por mutações no gene supressor de tumor NF1, localizado no cromossomo 17 (17q11.2), que codifica a neurofibromina (nf), uma proteína capaz de regular negativamente a via de sinalização Ras-Raf/MAPK que ativa a proliferação celular. Mutações no gene NF1 resultam em alteração ou perda de função do regulador negativo de crescimento e diferenciação celular de NF, que conduz à proliferação celular descontrolada e ao aumento do risco de desenvolvimento de câncer.4,5
Uma mulher de 26 anos com NF1 foi atendida na nossa instituição com dor intensa na região subungueal do quinto quirodáctilo direito, no hiponíquio e na prega lateral da unha do terceiro quirodáctilo esquerdo e na região subungueal do hálux esquerdo.
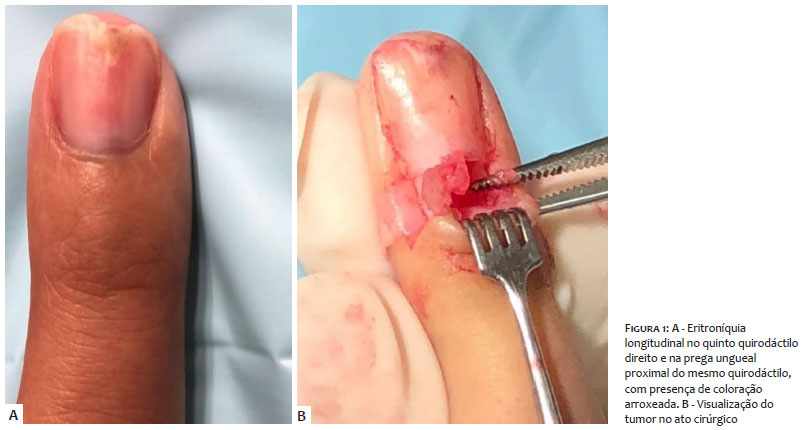
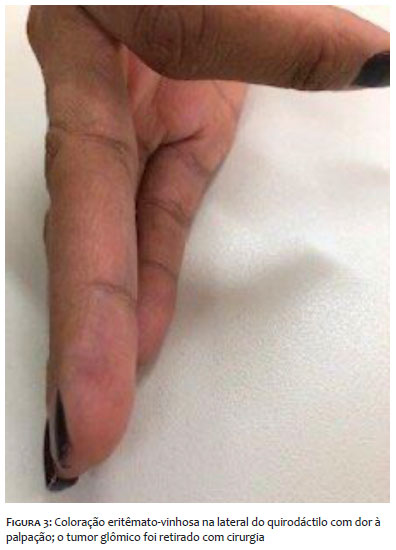
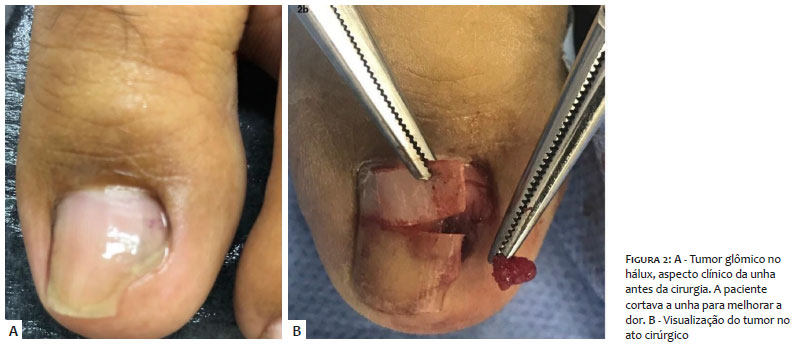
Clinicamente, apresentava eritroníquia longitudinal no quinto quirodáctilo direito e na prega ungueal proximal do mesmo quirodáctilo, coloração arroxeada e dor local (Figuras 1 e 3). O terceiro quirodáctilo não apresentava eritroníquia longitudinal, apenas dor local, enquanto o hálux esquerdo apresentava coloração purpúrica no meio da unha (Figura 2).
A paciente apresentava a tríade de Carroll (dor intensa, sensibilidade pontual e sensibilidade ao frio) em todos os dedos.
Um ultrassom de alta frequência mostrou um tumor de 6mm no quinto quirodáctilo direito.
Após anestesia troncular, foi feita incisão oblíqua na porção lateral de ambos os lados da prega proximal ungueal, expondo a matriz ungueal. Foram feitas avulsão da unha e incisão no leito ungueal, visualizando-se tumoração avermelhada, facilmente destacada por divulsão apenas com uma pinça mosquito.
O exame histológico do tumor confirmou o diagnóstico de tumor glômico.
Os tumores glômicos, geralmente presentes nos dedos das mãos e dos pés, são tumores benignos que se desenvolvem a partir de células que se assemelham às células musculares lisas modificadas dos aparelhos glômicos.6
A NF1, um distúrbio hereditário autossômico dominante, é causada por mutações do gene NF1 localizado no cromossomo 17. A proteína codificada pelo gene NF1, neurofibromina, atua como um supressor de tumor. Pacientes com NF1 perdem a expressão da neurofibromina, o que leva ao aumento da proliferação celular associada à proteína quinase, tendo propensão a desenvolver muitos tipos diferentes de tumores.7
O presente caso, com quatro tumores glômicos excisados, indica, juntamente com alguns casos relatados anteriormente, que existe uma associação entre os tumores glômicos e a NF1.
Em 1938, Klaber forneceu o primeiro relato de um tumor glômico surgido em um paciente com neurofibromatose.8 A literatura não contém mais relatos de tumores glômicos em pacientes com neurofibromatose até 1995, quando Sawada et al.3 relataram três pacientes com neurofibromatose tipo I e tumores glômicos subungueais. Entre 1995 e 2013, mais 13 relatos de casos descreveram tumores glômicos surgidos no cenário da neurofibromatose tipo I. Embora muitos dos autores tenham especulado sobre a possibilidade de uma associação, isso não foi confirmado até 20091, quando Brems et al.9 firmemente estabeleceram uma relação causal entre os tumores NF1 e glômicos: os tumores glômicos associados à NF1 exibem inativação bialélica do gene NF1, de modo que sete dos 12 tumores estudados abrigavam mutações gênicas e somáticas do gene NF1, enquanto dois tumores esporádicos glômicos não apresentavam anormalidades no gene NF1.
Em 2013, Harrison et al.2 realizaram um estudo epidemiológico de caso-controle no qual uma coorte de pacientes submetidos à excisão de tumor glômico foi comparada a uma coorte semelhante de pacientes que foram submetidos à excisão de outras lesões benignas da mão. Verificou-se que 29% dos pacientes submetidos à excisão do tumor glômico apresentavam o diagnóstico de neurofibromatose, enquanto nenhum dos pacientes da coorte controle apresentava o distúrbio.
A maioria dos tumores glômicos são únicos. É altamente improvável que haja tumores síncronos em dígitos adjacentes, como no presente caso. No entanto, em um paciente com NF1, o risco de tumores glômicos se eleva. Como ilustrado neste caso, o diagnóstico de tumores glômicos multifocais deve ser considerado nesses pacientes, especialmente quando a tríade clássica de características clínicas (dor espontânea paroxística, sensibilidade pontual e hipersensibilidade ao frio) está presente. A maioria está localizada na área subungueal. Diagnóstico e tratamento retardados podem levar à debilitação desnecessária em tais pacientes.10
O relato de caso fornece suporte adicional à noção de que NF1 tem um risco associado a múltiplos tumores glômicos. Para dermatologistas que gerenciam pacientes com NF1, o conhecimento dessa associação pode facilitar o diagnóstico precoce e o tratamento adequado.
Marcia Ferraz Nogueira | 0000-0001-7872-7304
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Jayme de Oliveira Filho | 0000-0003-0239-0981
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; participação efetiva na orientação da pesquisa.
Gabriela Machado Dias Junqueira | 0000-0003-0899-9341
Concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura.
Ana Julia Marçal Pereira Dias | 0000-0002-7063-3298
Concepção e planejamento do estudo; elaboração e redação do manuscrito; participação intelectual em conduta propedêutica e/ ou terapêutica de casos estudados; revisão crítica da literatura.
1. Harrison B. Tumores Sammer D. Glomus e Neurofibromatose: uma associação recentemente reconhecida. Open Glob Cir Plást Reconstr. 2014;2(9):e214.
2. Harrison B, Moore AM, Calfee R, et al. A associação entre tumores glômicos e neurofibromatose. J Hand Surg Am. 2013;38:1571-4.
3. Huson S. Neurofibromatose: fenótipos emergentes, mecanismos e a gestão. Clin Med. 2008;8(6):611.
4. Scaravilli G, Rossi R, Artiaco S, Merolla G. Glomus tumor da eminência tenar na neurofibromatose tipo 1: relato de caso e revisão de literatura. Med Transl UniSa . 2015;11:63-8.
5. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Diretrizes para o diagnóstico e tratamento de indivíduos com neurofibromatose 1. J Med Genet. 2007;44(2):81-8.
6. Weedon D. Glomus tumor. In: Weedon D, editor. Patologia da pele de Weedon, 3th ed. China: Churchill Livingstone Elsevier; 2010. p. 907-8.
7. Brems H, Beert E, Ravel T, Legius E. Mecanismos na patogênese de tumores malignos na neurofibromatose tipo 1. Lancet Oncol. 2009;10:508-15.
8. Klaber R. Morbus recklinghausen com tumores glomóides. Proc R Soc Med. 1938; 31:347.
9. Brems H, Park C, Maertens O, Pemov A, Messiaen L, Upadhyaya M, et al. Tumores glômicos na neurofibromatose tipo 1: evidência genética, funcional e clínica de uma nova associação. Cancer Res 2009;69:7393-401.
10. Aqil N, Gallouj S, Moustaide K, Mernissi F. Tumores dolorosos em um paciente com neurofibromatose tipo 1: relato de caso. J Med Case Rep. 2018;12:319.
 All content the journal, except where identified, is under a Creative Commons Attribution-NonCommercial 4.0 International license - ISSN-e 1984-8773
All content the journal, except where identified, is under a Creative Commons Attribution-NonCommercial 4.0 International license - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}