


Debora Pedroso de Almeida1; Lissa Sabino de Matos Wegher1; Diogo Hiroshi Mizumoto1; André Luiz Simião2
Received on: 20/05/2020
Approved on: 15/11/2020
Financial support: None
Conflict of interest: None
Study conducted at the Dermatology Service of the Pontifícia Universidade Católica de Campinas, Campinas (SP), Brazil
We report the case of a 13-year-old girl with a diagnosis of extra-abdominal desmoid fibromatosis, a neoplasm of benign origin but uncommon in the soft tissues. This study aims to expose the rarity of the tumor and its challenging therapeutic approach due to its high frequent local recurrence rates. For the treatment, Mohs micrographic surgery was performed in two stages to obtain a free margin.
Keywords: Fibromatosis, Aggressive; Mohs Surgery; Neoplasms
Extra-abdominal desmoid fibromatosis is a rare, benign neoplasm, originating in fibroblastic cells and growing in a variable pattern. It can occur in almost any part of the body. Despite not presenting malignant behavior, such as metastases, the desmoid tumor has a high capacity for growth and local invasion. With few cases described in the literature, its surgical treatment is practically a consensus.1,2,3 The Mohs micrographic surgery (MMS) approach considerably reduces the chances of frequent recurrence of these tumors, estimated at approximately 11% to 64%, depending on the treatment used.4 We report a pediatric patient’s case, with this neoplasm located on the lower lip, submitted to excision by micrographic surgery and followed for two years, without recurrence.
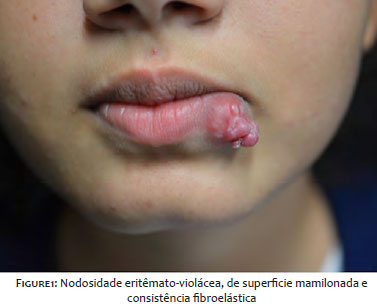
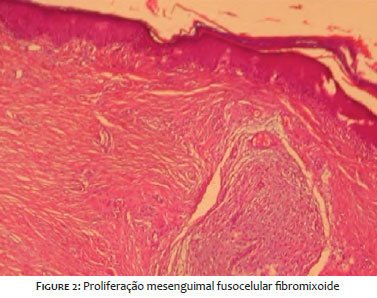
A 13-year-old girl presented a history of a lesion on the lower lip beginning seven months ago, with slow and progressive growth. She denied any symptoms or previous trauma at the site. The patient had a personal history of juvenile idiopathic arthritis (JIA), and she was using methotrexate, folic acid, and etanercept. The dermatological examination showed an erythematous-violaceous tumor, with a nipple-like surface and a well-defined fibroelastic consistency of approximately 2.5 cm on the lower lip, on the left Figure 1. An excisional biopsy was performed, revealing fibromyxoid fusocellular mesenchymal proliferation, with an associated inflammatory cell component, on anatomopathological examination Figure 2. The immunohistochemical panel showed diffuse and intense positivity of beta-catenin and vimentin and negative ALK-1. These findings pointed to the diagnosis of extra-abdominal desmoid fibromatosis.5
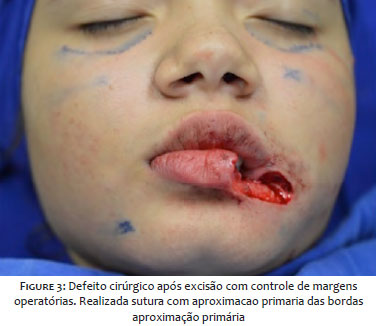
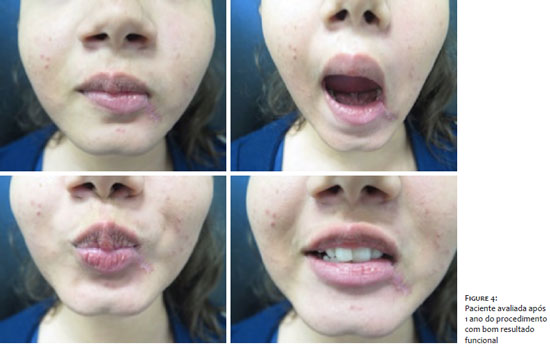
We opted for the excision of the lesion by Mohs micrographic surgery, revealing compromised margins in the first stage of the procedure and free margins after the surgical defect’s enlargement Figure 3. The patient was followed every three months in the first year and every six months in the second, with no clinical sign of recurrence of the lesionFigure 4 .
The desmoid tumor or desmoid fibromatosis consists of fibroblastic proliferation, originating in the soft tissues, with benign behavior. McFarlane first described it in 1832, and Muller coined the term desmoid five years later, based on the myofibroblastic cells that constitute these tumors. It has an estimated frequency of 3% of all soft tissue tumors. It most commonly affects individuals between 15 and 60 years old, being rare in the pediatric age group and after the fourth decade, with a slight preference for women. Its most frequent form of presentation is intra-abdominal, accounting for about 70% of cases.6,7,8
The vast majority of cases, around 90%, are sporadic. Somatic mutations of the beta-catenin protein, which makes up the CTNNB1 gene, may boost this tumor’s development.9 In addition to the genetic characteristics, other endocrine and physical factors, such as trauma, play an essential role in the disease’s etiology.
In its rare, extra-abdominal form, its appearance in the extremities’ soft tissues is more frequently observed, with half of the cases originating in the limbs, 43% in the chest, and only 7% in the head and neck region. However, any anatomical area is subject to its appearance. 10,11,12 Our report stands out for its atypical presentation, both by the age group and by its very unusual location.
These tumors are characterized by presenting different degrees of aggressiveness with unpredictable biological behavior. Their natural history ranges from indolent and self-limited lesions to infiltrative lesions and with rapid local proliferation. The symptomatology depends on the presentation site and adjacent structures. The majority is asymptomatic; however, due to its significant regional growth, it can compress or compromise other structures, organs, or the functionality of the affected region.13,14
The diagnosis is made by anatomopathological examination that shows bundles of spindle cells, elongated, in the middle of stromal collagen, with variable vascularization. The cells are usually small, with clear cytoplasm and pale nuclei, showing no atypia or mitoses. Immunohistochemistry is positive for muscle cell markers, such as vimentin, desmin, and smooth muscle actin. In electron microscopy, spindles cells look like myofibroblasts.
The most common cases, intra-abdominal, usually require imaging methods to complement the diagnosis.7
According to most authors, the ideal treatment has not yet been established, although surgery is the first therapeutic option. The main challenge is the fact that, despite being histologically benign, they have high recurrence rates. Most of the cases described in the literature consist of intra-abdominal tumors where conservative treatment can be considered. However, there are few publications of lesions outside the abdominal cavity and in pediatric patients. Nevertheless, when conventional deciding for local resection, it must contain a wide margin to avoid the risk of incomplete excision.6 Other therapeutic options include chemotherapy, hormone therapy, and radiation therapy.
In the case reported, the surgical approach, especially the treatment with MMS, was strictly necessary, considering the patient’s age, the unfavorable anatomical location in a prime area, and the relevant percentage of these tumors’ recurrence. Two surgical stages were necessary to obtain the free margins.
Health professionals must remember the desmoid tumor as a differential diagnosis of soft tissue tumors, and anatomopathological examination is essential for its diagnostic confirmation. The effectiveness of MMS is already widespread and should be indicated for the removal of tumors with potential for recurrence and in risk areas where tissue preservation is essential, as in our case. This approach was fundamental in healing, ensuring margin control and excellent aesthetic and functional results for the patient.
Debora Pedroso de Almeida | 0000-0001-6543-5898 Study design and planning; preparation and writing of the manuscript; data collection, analysis, and interpretation; critical literature review.
André Luiz Simião | 0000-0002-0246-2001 Approval of the final version of the manuscript; critical revision of the manuscript.
Lissa Sabino de Matos Wegher | 0000-0002-3247-3393 Study design and planning; critical revision of the manuscript.
Diogo Hiroshi Mizumoto | 0000-0002-4979-3959 Study design and planning; critical literature review.
1. Meazza C, Bisogno G, Gronchi A, Fiore M, Cecchetto G, Alaggio R, et al. Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer 2010;116(1):233-40.
2. Reitamo JJ, Scheinin TM, Hayry P. The desmoid syndrome: new aspects in the cause, pathogenesis and treatment of the desmoids tumor. Am J Surg. 1986;151(2):230-7.
3. van Broekhoven DLM, Verhoef C, Elias SG, Witkamp AJ, van Gorp JMHH, van Geel BAN, et al. Local recurrence after surgery for primary extra-abdominal desmoid-type fibromatosis. Br J Surg 2013;100(9):1214-9.
4. Mueller C, Croner R., Klein P, Grützmann R, Vassos, N. Primary and recurrent sporadic desmoids: Prognostic factors influencing recurrence-free survival after complete gross resection. Int J Surg. 2016;31:63-70.
5. Shen C, Wang C, Yan J, He T, Zhou X, Ma W, Zhang B. Clinicopathological characteristics, treatment, and survival outcomes of retroperitoneal desmoid-type fibromatosis. Medicine (Baltimore). 2019;98(47):e18081.
6. Teixeira LEM, Arantes EC, Villela RF, Soares CBG, Costa RBC, Andrade MAP, et al. Extra abdominal desmoid tumor: local recurrence and treatment options. Acta Ortop Bras. 2016;24(3):147-50.
7. Valejo FAM, Tiezzi DM, Nai GA. Tumor desmoide abdômino-pélvico. Rev Bras Ginecol Obstet. 2009;31(1):35-40.
8. Reitamo JJ, Hayry P, Nykyri E, Sax ¨ en E. The desmoid tumour. I. Incidence, sex- age- and anatomical distribution in the Finnish population. Am J Clin Pathol. 1982;77(6):665-73.
9. Mullen JT, DeLaney TF, Rosenberg AE, Le L, Iafrate AJ, Kobayashi W, et al. b-Catenin mutation status and outcomes in sporadic desmoid tumors. Oncologist 2013;18(9):1043-9.
10. Rock MG, Pritchard DJ, Reiman HM, Soule EH, Brewster RC. Extra-abdominal desmoid tumors. J Bone Joint Surg Am. 1984;66(9):1369-74.
11. Siva AF, Alves JCRR, Portugal EH, Fonseca RPL, Almeida ACM, Pereira NA, et al. Fibromatose agressiva (tumor desmoide) associada ao implante mamário: revisão da literatura e apresentação de três novos casos. Rev Bras Cir Plást. 2017;32(3):361-71.
12. Merchant NB, Lewis JL, Woodruff JM, Leung DH, BrennanMF. Extremity and trunk desmoid tumors. A multifactorial analysis of outcome. Cancer. 1999;86 (10):2045-52.
13. Shields CJ, Winter DC, Kirwan WO, Redmond HP. Desmoid tumours. Eur J Surg Oncol. 2001;27(8):701-6.
14. Fallen T, Wilson M, Morlan B, Lindor NM. Desmoid tumors e a characterization of patients seen at Mayo Clinic 1976-1999. Fam Cancer. 2006;5(2):191-4
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}