


Debora Pedroso de Almeida1; Lissa Sabino de Matos Wegher1; Diogo Hiroshi Mizumoto1; André Luiz Simião2
Data de recebimento: 20/05/2020
Data de aprovação: 15/11/2020
Trabalho realizado no Serviço de Dermatologia da Pontifícia Universidade Católica de Campinas, Campinas (SP), Brasil.
Suporte Financeiro: Nenhum.
Conflito de Interesses: Nenhum.
Apresenta-se caso de paciente do gênero feminino, 13 anos, com diagnóstico de fibromatose desmoide extra-abdominal, neoplasia dos tecidos moles, de origem benigna, porém incomum. Nosso objetivo é expor este relato pela raridade do tumor bem como por sua desafiadora abordagem terapêutica e pelas altas taxas de recidiva local frequentes nesses tumores. Para o tratamento, realizou-se cirurgia micrográfica de Mohs em duas fases para obtenção de margens livres.
Keywords: Cirurgia de Mohs; Fibromatose Agressiva; Neoplasias
A fibromatose desmoide extra-abdominal é uma neoplasia rara, benigna, com origem nas células fibroblásticas e crescimento de padrão variável. Pode ocorrer em praticamente qualquer parte do corpo e, apesar de não apresentar comportamento maligno, como metástases, o tumor desmoide tem uma alta capacidade de crescimento e invasão local. Com poucos casos descritos na literatura, seu tratamento cirúrgico é praticamente um consenso.1,2,3 A abordagem pela cirurgia micrográfica de Mohs (CMM) reduz consideravelmente as chances de recidiva frequentes desses tumores, estimada em aproximadamente 11 até 64%, dependendo do tratamento empregado.4 Relatamos um caso de paciente na faixa etária pediátrica, com esta neoplasia localizada no lábio inferior, submetida à exérese pela cirurgia micrográfica e acompanhada por período de dois anos, sem recorrência.
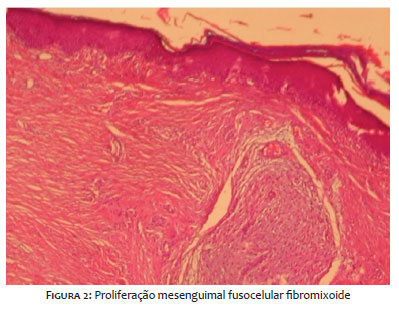
Paciente do gênero feminino, 13 anos, com antecedente pessoal de artrite idiopática juvenil, em uso de metotrexato, ácido fólico e etanercepte. Apresentava história de lesão no lábio inferior de início há sete meses, com crescimento lento e progressivo. Negava qualquer sintomatologia ou trauma prévio no local. Ao exame dermatológico, apresentava tumoração eritêmato-violácea, de superfície mamilonada e consistência fibroelástica, bem delimitada, de aproximadamente 2,5cm no lábio inferior, à esquerda (Figura1). Foi realizada biópsia excisional que evidenciou, ao exame anatomopatológico, proliferação mesenquimal fusocelular fibromixoide, com componente de células inflamatórias associado (Figura 2). O painel imuno-histoquímico revelou beta catenina e vimentina positivo difuso intenso e ALK-1 negativo, achados que apontam para o diagnóstico de fibromatose desmoide extra-abdominal.5
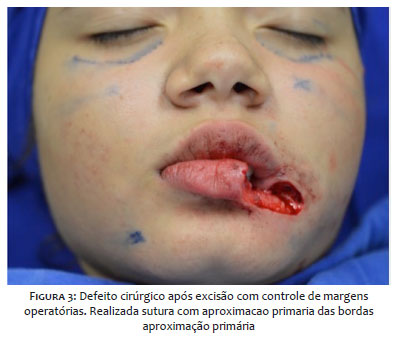
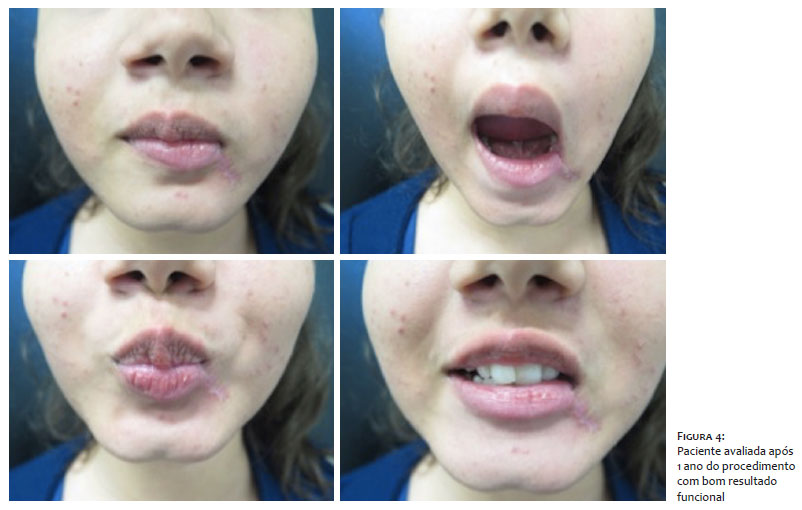
Optou-se pelo tratamento com exérese da lesão pela cirurgia micrográfica de Mohs, revelando margens comprometidas no primeiro estágio do procedimento, e livres de neoplasia após a ampliação do defeito cirúrgico (Figura 3). A paciente foi seguida trimestralmente no primeiro ano e semestralmente no segundo, sem sinal clínico de recidiva da lesão (Figura 4).
O tumor desmoide ou fibromatose desmoide consiste em proliferação fibroblástica, de origem nos tecidos moles, com comportamento benigno. Foi descrito pela primeira vez em 1832 por McFarlane e o termo desmoide foi instituído cinco anos depois por Muller, com base nas células miofibroblásticas que compõem esses tumores. Apresenta uma frequência estimada de 3% de todos os tumores de tecidos moles. Com discreta predileção pelo gênero feminino, acomete mais comumente indivíduos entre 15 e 60 anos, sendo raro na faixa etária pediátrica e após a quarta década. Sua forma mais comum de apresentação é intra-abdominal, sendo responsável por cerca de 70% dos casos.6,7,8
A grande maioria dos casos, em torno de 90%, é esporádica. Acredita-se que mutações somáticas da proteína beta catenina, que compõe o gene CTNNB1, possam impulsionar o desenvolvimento deste tumor.9 Além do fator genético, outros fatores endócrinos e físicos, como trauma, desempenham papel importante na etiologia da doença.
Na sua forma rara, extra-abdominal, é mais frequentemente observado o seu surgimento nos tecidos moles das extremidades, sendo que metade dos casos tem origem nos membros, 43% no tórax e somente 7% na região de cabeça e pescoço. No entanto, qualquer região anatômica está sujeita ao seu aparecimento. 10,11,12 O nosso relato destaca-se pela apresentação atípica, tanto pela faixa etária quanto por sua localização bastante incomum.
Esses tumores são caracterizados por apresentarem distintos graus de agressividade com comportamento biológico imprevisível, sendo que sua história natural varia desde lesões indolentes e autolimitadas a lesões infiltrativas e com rápida proliferação local. A sintomatologia depende do local de apresentação e estruturas adjacentes. A maioria é assintomática, porém, pelo seu significante crescimento regional, pode comprimir ou comprometer outras estruturas, órgãos ou a funcionalidade da região acometida.13,14
O diagnóstico é realizado pelo exame anatomopatológico que evidencia feixes de células fusiformes, alongadas, em meio a estroma de colágeno, com vascularização variável. As células geralmente são pequenas, com citoplasma claro e núcleos pálidos, não apresentando atipias ou mitoses. A imuno-histoquímica é positiva para marcadores de células musculares, como vimentina, desmina e actina de músculo liso. Na microscopia eletrônica, as células em fusos se parecem com miofibroblastos.
Nos casos mais comuns, intra-abdominais, geralmente se fazem necessários métodos de imagem para complementar o diagnóstico.7
O tratamento ideal ainda não foi estabelecido, embora a cirurgia seja a primeira opção terapêutica, segundo a maioria dos autores. A principal dificuldade está no fato de que, apesar de serem histologicamente benignos, possuem elevadas taxas de recorrência. A maioria dos casos descritos na literatura consiste em tumores intra-abdominais em que o tratamento conservador pode ser considerado. Porém, existem poucas publicações de lesões fora da cavidade abdominal e em pacientes pediátricos. No entanto, quando decidida a ressecção local convencional, esta deve conter margem ampla, de forma a evitar o risco de excisão incompleta.6 Outras opções terapêuticas descritas incluem quimioterapia, terapia hormonal e radioterapia.
No caso relatado, a abordagem cirúrgica, em especial o tratamento com a CMM, fez-se estritamente necessária, considerando-se a idade da paciente, a localização anatômica desfavorável em área nobre e o percentual relevante de recorrência desses tumores. Foram necessários dois estágios cirúrgicos para a obtenção das margens livres.
O tumor desmoide deve ser lembrado como diagnóstico diferencial de tumorações de partes moles, e o exame anatomopatológico é essencial para sua confirmação diagnóstica. A eficácia da CMM já é amplamente difundida e deve ser indicada para a remoção de tumores com potencial de recidiva e em áreas de risco em que a preservação tecidual é essencial, como no caso demonstrado. Essa abordagem foi fundamental na cura, garantindo o controle das margens e o excelente resultado estético e funcional para a paciente.
Debora Pedroso de Almeida | 0000-0001-6543-5898
Concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura.
André Luiz Simião | 0000-0002-0246-2001
Aprovação da versão final do manuscrito; revisão crítica do manuscrito
Lissa Sabino de Matos Wegher | 0000-0002-3247-3393
Concepção e planejamento do estudo; revisão crítica do manuscrito.
Diogo Hiroshi Mizumoto | 0000-0002-4979-3959
Concepção e planejamento do estudo; revisão crítica da literatura.
1. Meazza C, Bisogno G, Gronchi A, Fiore M, Cecchetto G, Alaggio R, et al. Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer 2010;116(1):233-40.
2. Reitamo JJ, Scheinin TM, Hayry P. The desmoid syndrome: new aspects in the cause, pathogenesis and treatment of the desmoids tumor. Am J Surg. 1986;151(2):230-7.
3. van Broekhoven DLM, Verhoef C, Elias SG, Witkamp AJ, van Gorp JMHH, van Geel BAN, et al. Local recurrence after surgery for primary extra-abdominal desmoid-type fibromatosis. Br J Surg 2013;100(9):1214-9.
4. Mueller C, Croner R., Klein P, Grützmann R, Vassos, N. Primary and recurrent sporadic desmoids: Prognostic factors influencing recurrence-free survival after complete gross resection. Int J Surg. 2016;31:63-70.
5. Shen C, Wang C, Yan J, He T, Zhou X, Ma W, Zhang B. Clinicopathological characteristics, treatment, and survival outcomes of retroperitoneal desmoid-type fibromatosis. Medicine (Baltimore). 2019;98(47):e18081.
6. Teixeira LEM, Arantes EC, Villela RF, Soares CBG, Costa RBC, Andrade MAP, et al. Extra abdominal desmoid tumor: local recurrence and treatment options. Acta Ortop Bras. 2016;24(3):147-50.
7. Valejo FAM, Tiezzi DM, Nai GA. Tumor desmoide abdômino-pélvico. Rev Bras Ginecol Obstet. 2009;31(1):35-40.
8. Reitamo JJ, Hayry P, Nykyri E, Sax ¨ en E. The desmoid tumour. I. Incidence, sex- age- and anatomical distribution in the Finnish population. Am J Clin Pathol. 1982;77(6):665-73.
9. Mullen JT, DeLaney TF, Rosenberg AE, Le L, Iafrate AJ, Kobayashi W, et al. b-Catenin mutation status and outcomes in sporadic desmoid tumors. Oncologist 2013;18(9):1043-9.
10. Rock MG, Pritchard DJ, Reiman HM, Soule EH, Brewster RC. Extra-abdominal desmoid tumors. J Bone Joint Surg Am. 1984;66(9):1369-74.
11. Siva AF, Alves JCRR, Portugal EH, Fonseca RPL, Almeida ACM, Pereira NA, et al. Fibromatose agressiva (tumor desmoide) associada ao implante mamário: revisão da literatura e apresentação de três novos casos. Rev Bras Cir Plást. 2017;32(3):361-71.
12. Merchant NB, Lewis JL, Woodruff JM, Leung DH, BrennanMF. Extremity and trunk desmoid tumors. A multifactorial analysis of outcome. Cancer. 1999;86 (10):2045-52.
13. Shields CJ, Winter DC, Kirwan WO, Redmond HP. Desmoid tumours. Eur J Surg Oncol. 2001;27(8):701-6.
14. Fallen T, Wilson M, Morlan B, Lindor NM. Desmoid tumors e a characterization of patients seen at Mayo Clinic 1976-1999. Fam Cancer. 2006;5(2):191-4
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}