


Anna Carolina Brandão Vasconcelos; John Verrinder Veasey
Received on: 29/08/2019
Approved on: 18/08/2020
Financial support: None
Conflict of interest: None
Study conducted at the Dermatology Clinic of the Hospital da Santa Casa de São Paulo, São Paulo (SP), Brazil
Dowling-Degos disease (DDD) is a rare genodermatosis. Progressive reticular hyperpigmentation of flexural areas mainly characterizes the disease, which may be associated with a large spectrum of benign lesions and cutaneous neoplasms. It may cause psychosocial impairment due to the deformity caused by the lesions with significant aesthetic damage. We present two cases of male patients with hyperchromic macules, multiple comedones, epidermal cysts, cribriform scars on the face, cervical region, dorsum, anterior thorax, armpits, genital region, and disfiguring tumors, characteristic of DDD.
Keywords: Carcinoma; Genetics; Hidradenitis; Hidradenitis Suppurativa; Skin Pigmentation
Dowling-Degos disease (DDD) is a rare genodermatosis, with an autosomal dominant transmission, variable penetrance, and expressiveness, initially described by Jones and Grice in 1974.1,2
It is a late-onset disease, after the second decade of life. Reticulated dyschromia, mainly on the face and flexural surfaces such as neck, armpits, elbow pit, submammary areas, and groins, characterize the condition.3,4 Concomitantly, lesions of the pilosebaceous unit may occur, such as comedones, epidermal cysts, abscesses, hidradenitis suppurativa, in addition to skin neoplasms, such as squamous cell carcinoma and keratoacanthoma.2,4 Pigmentation is progressive and symmetrical, usually extensive and asymptomatic, exacerbated by sun exposure.5,6,7
Differential diagnoses should be made with acanthosis nigricans, acropigmentation of Kitamura, Galli-Galli disease, dyschromatosis universalis hereditaria, and dyschromatosis symmetrica hereditaria.8 Such conditions may present clinical overlaps with each other, and some authors can consider them as different diseases, while others, the spectrum of the same disease.4,5,9
The diagnosis is based on suggestive clinical characteristics associated with the findings on histopathological examination.7,8 Histopathology shows hyperpigmentation of the basal layer, filiform proliferation of the epidermis, sometimes similar to reindeer horn, in addition to hyperkeratosis and budding, arising from the hair infundibulum, featuring a follicular plug.1,3,6 Perivascular lymphohistiocytic infiltrate in the papillary dermis and horny pseudocysts can be observed, with a standard number of melanocytes.7,10 Genetic tests reveal mutations in keratin 5 (KRT5), protein O-glycosyltransferase 1 (POGLUT1), protein O-fucosyltransferase 1 (POFUT1), and PSENEN gene as causing the disease. Patients with the latter mutation present lesions of hidradenitis suppurativa.2,4
There is no definitive treatment for DDD.7 The treatments are unsatisfactory, and topical hydroquinone, tretinoin, adapalene, and corticosteroids can be used, in addition to Er:YAG laser, aiming to reduce the risk of post-inflammatory hyperpigmentation.1,7 Isotretinoin would be an option since the condition promotes a change in keratinization.1 Cystic and tumoral lesions should be treated with surgical excision.7
We conducted a retrospective descriptive study of two cases diagnosed with Dowling-Degos disease at the Dermatology Clinic from November 2016 to May 2019.
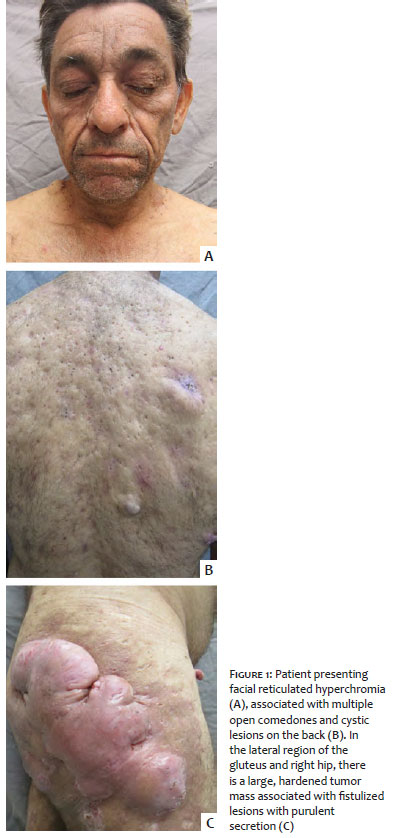
A 58-year-old man reported hyperpigmentation on the face, cervical region, and flexural areas for ten years, associated with the progressive appearance of multiple cysts, which drained purulent exudate, and a tumor on the buttock and lateral aspect of the right thigh. On clinical examination, the patient had predominant hyperpigmentation on the face and flexural areas, multiple open comedones, and epidermoid cysts distributed throughout the body and on the hips and buttocks, hidradenitis suppurativa-like lesions, and a keloid-like tumor (Figure 1). He denied similar cases in the family.
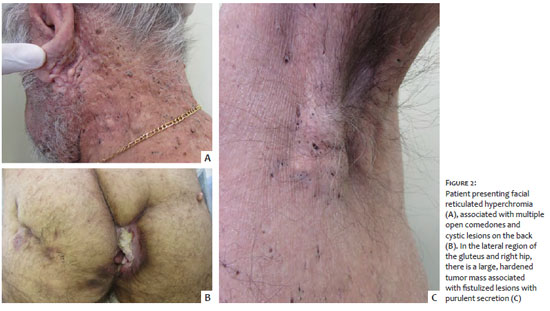
A 59-year-old man presented suppurative lesions in the gluteal region associated with progressive growth tumor for one year. On clinical examination, he showed dermatosis characterized by multiple comedones spread throughout the body, associated with hidradenitis suppurativa-like lesions in the left buttocks, and vegetative and ulcerated tumor in the perianal and intergluteal region, whose biopsy diagnosed invasive squamous cell carcinoma (Figure 2). He denied similar cases in the family.
DDD is a late-onset genodermatosis, usually in adulthood, which initially affects the armpits and groins, and later, the intergluteal and inframammary regions, neck, and trunk.1 Both patients presented the condition in adulthood, and the lesions clearly show the preference for these locations. Although this disease predominantly affects women in a proportion of 2:1,1 we present two male patients.
The patients reported here presented not only the dyschromic manifestations of the syndrome, but also several alterations in the pilosebaceous unit spread throughout the body, from open comedones to hidradenitis, with a high impact on the quality of life, in general, and on self-esteem, in particular, including significant aesthetic changes.
The exuberance of the lesions in this study demonstrates why this disease is also called the “dark dot disease” (DDD) by some authors.1 Despite not having inflammatory phenomena in their benign evolution, these lesions are deeply unsightly. In the second patient, we also observed intergluteal squamous cell carcinoma, a neoplasm reported in association with this disease.2,4 In addition to both cases’ clinical aspect characteristics, a histopathological examination confirmed the diagnoses, which was compatible with the findings described in the literature.
Finally, it is essential to note that the present study was conducted at the Dermatology Clinic of a tertiary reference hospital in the country’s largest city. In three years of care, only two cases of DDD were evidenced, showing the rarity of this dermatosis in the general population.
Anna Carolina Brandão Vasconcelos | 000-0002-2171-4358
Approval of the final version of the manuscript; study design and planning; preparation and writing of the manuscript; data collection, analysis, and interpretation; active participation in research orientation; intellectual participation in propaedeutic and/or therapeutic conduct of studied cases; critical literature review; critical revision of the manuscript.
John Verrinder Veasey | 0000-0002-4256-5734
Approval of the final version of the manuscript; study design and planning; preparation and writing of the manuscript; active participation in research orientation; intellectual participation in propaedeutic and/or therapeutic conduct of studied cases; critical literature review; critical revision of the manuscript.
1. Zimmermann CC, Sforza D, Macedo PM, Azulay-Abulafia L, Alves MFGS, Carneiro SCS. Doença de Dowling-Degos: apresentação clínica e histopatológica clássica. An Bras Dermatol. 2011;86(5):979-82.
2. Fenske NA, Groover CE, Lober CW, Espinoza CG. Dowling-Degos disease, hidradenitis suppurativa, and multiple keratoacanthomas: a disorder that may be caused by a single underlying defect in pilsebaceous epithelial proliferation. J Am Acad Dermatol. 1991;24(5 Pt 2):888-92.
3. Kim YC, Davis MD, Schanbacher CF, Su WP. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999;40(3):462-7.
4. Linke M, Orouji A, Géraud C. Vesicular variant of Dowling-Degos disease. Br J Dermatol. 2018;179(3):795-6.
5. Ujihara M, Kamakura T, Ikeda M, Kodama H. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147(3):568-71.
6. Bhagwat PV, Tophakhane RS, Shashikumar BM, Noronha TM, Naidu V. Three cases of Dowling Degos disease in two families. Indian J Dermatol Venereol Leprol. 2009;75(4):398-400.
7. Hohmann CB, Koche B, Bonamigo RR, Dornelles ST, Cattani CAS. Caso para diagnostico. Doenca de Dowling-Degos e ceratoacantoma. An Bras Dermatol. 2010;85(2):241-3.
8. Gontijo B. O espectro doença de Kitamura - doença de Dowling-Degos. An Bras Dermatol. 1993;68(6):89-92.
9. Wu YH, Lin YC. Generalized Dowling-Degos disease. J Am Acad Dermatol. 2007;57(2):327-34.
10. Rathoriya SG, Soni SS, Asati D. Dowling-Degos disease with reticulate acropigmentation of Kitamura: extended spectrum of a single entity. Indian Dermatol Online J. 2016;7(1):32-5.
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}