


Fernanda Guimarães Souza1; Daniela Pessanha dos Santos1; Luiza Oliveira Ribeiro1; Thiago Rubim Batista Bellot Nascimento2; Flávio Barbosa Luz3
Financing: None.
Conflicts of interest: None.
Submitted on: 02/26/2024
Final decision: 11/21/2024
How to cite this article: Souza FG, Santos DP, Ribeiro LO, Nascimento TRBB, Luz FB. Ontogenisis of Merkel-cell carcinoma. Surg Cosmet Dermatol. 2024;16:e202040354.
A narrative review was performed to clarify the ontogenesis of Merkel-cell carcinoma. It is hypothesized that the cells are derived from the embryonic neural crest, skin stem cells, epithelial cells, pre-/pro-B cells, dermal fibroblasts, or metastatic cells from other types of carcinomas. The influence of epigenetics was also considered. Methodologically, to prepare the summary articles were selected from the PubMed and SciELO databases and cited references. The results suggest that it is more likely that Merkel-cell carcinoma has multiple origins, possibly from poorly differentiated cells.
Keywords:
A narrative review was performed to clarify the ontogenesis of Merkel-cell carcinoma. It is hypothesized that the cells are derived from the embryonic neural crest, skin stem cells, epithelial cells, pre-/pro-B cells, dermal fibroblasts, or metastatic
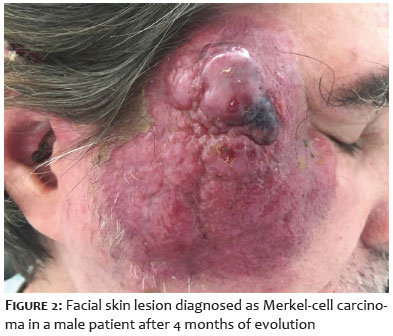
Merkel-cell carcinoma (MCC), formerly known as trabecular carcinoma of the skin, is a rare and aggressive carcinoma with neuroendocrine characteristics, high mortality, and frequent metastasis. It presents as a firm, painless, rapidly growing skin lesion with a smooth surface and erythematous-violet coloration (Figure 1).1 The tumor develops initially in the dermis, often progressing to the subcutaneous tissue, leading to metastasis through the lymphatic system. These characteristics explain its rapid spread and highlight the importance of early diagnosis. The main risk factors for this type of cancer are: advanced age, fair skin, exposure to ultraviolet radiation, and a compromised immune system. Furthermore, it is known that approximately 80% of MCC cases are associated with MC polyomavirus (MCPyV), a non-enveloped circular DNA virus belonging to the Polyomaviridae family that commonly infects skin keratinocytes. Most of the population first comes into contact with MCPyV during early childhood, and most infections are asymptomatic, with the virus remaining latent throughout life. Later, when poorly controlled by the immune system, it can give rise to virus-positive MCC. The remainder of MCC cases unrelated to MCPyV (virus-negative MCC) are caused primarily by mutations resulting from chronic exposure to ultraviolet radiation in the cells of tumor origin.2 The lesions share neuroendocrine markers with MCs, such as cytokeratin 20 and neuron-specific enolase, which led to their name.2 However, despite the apparent relationship, it is still unknown which cell line gives rise to this carcinoma. Since MCs are post-mitotic cells, they have limited oncogenic potential and are also located in the epidermis, factors which weaken the hypothesis that MCC originates from this cell type. Furthermore, tests indicate that MCs do not respond effectively to oncogenic stimuli, including polyomavirus antigens.3 It has been proposed that the following cell lines may give rise to MCC: cells derived from the embryonic neural crest, progenitor cells or skin stem cells, epithelial cells, pre- and pro-B cells, dermal fibroblasts close to hair follicles, or metastatic cells from other types of carcinomas.2-4 Some studies have also proposed that virus-negative MCCs originate from different cell lines and converge into the same phenotype through epigenetic reprogramming.2 Research aimed at defining the oncogenesis of MCC is extremely important for establishing specific biomarkers. This will enable medical advances to more appropriately treat MCC, such as prophylactic measures to reduce the incidence of the disease and improve prognosis, reducing the rates of metastasis and recurrence, as well as the number of deaths.
We conducted a narrative review based on 57 articles. A search was conducted of the PubMed and SciELO databases for studies published between 2008 and 2022 in English or Portuguese. The search terms were: "Merkel Cell Carcinoma" and "Origin". Of the 74 articles found, 42 that addressed the theoretical bases of the main ontological hypotheses of MCC were selected. Fifteen further publications cited in the selected articles were also included, totaling 57 articles. Finally, based on the selected material, a narrative review of the possible cellular origins of MCC was produced.
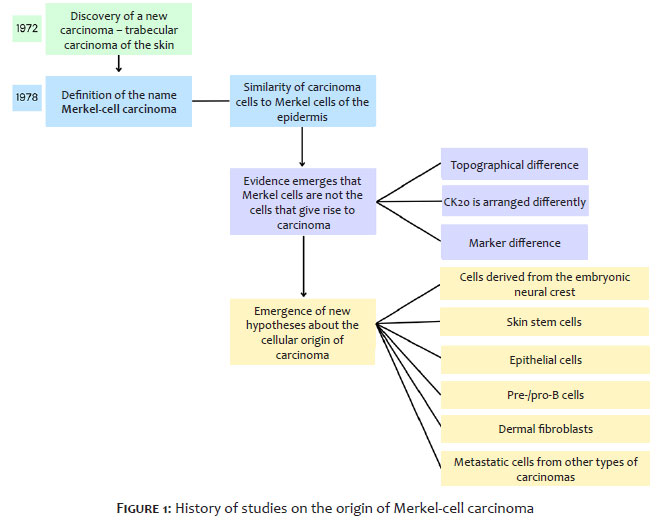
MCC was initially described in 1972 as trabecular carcinoma of the skin and, at that time, it was believed to be of eccrine origin.5 The name "Merkel-cell carcinoma" was applied in 1978 after immunohistochemical identification of the possible relationship between this carcinoma and MCs in the epidermis, as shown in Table 1. MCs, epidermal sensory cells that interact with low-threshold mechanoreceptor Aβ type 1 afferent neurons, are considered important in light touch perception.6 The hypothesis linking the newly discovered neoplasm to MCs was based on by their phenotypic similarities. Electron microscopy studies on trabecular carcinomas of the skin identified neurosecretory granules, which reinforced the theory that this neoplasm's origin could be MCs.7 However, later studies found several indications that it was unlikely that MCs were the origin of this carcinoma. Evidence supporting this new perspective included the topographical difference between MCCs and MCs: MCs are present in the epidermis, the most superficial layer of the skin, while MCCs are mostly found in the dermis.8 Furthermore, although both sets of cells express similar specific cytokeratins, such as cytokeratin 20, histological analysis indicates that these molecules are arranged differently in each cell type. In MCs, cytokeratin 20 is loosely organized, while in MCCs it is organized in spiral or plate-like arrangements.9 Data indicating that MCCs express markers not identified in MCs, such as the tyrosine kinase receptor c-kit and the adhesion molecule CD171, should also be considered.10 Thus, the question of which cell type is responsible for MCC has once again come to the fore. Neural crest cells were initially considered, since the pathogenic mechanism of MCC formation is similar to that of other neoplasms originating from these cells.11 The theory also emerged that pre-B and pro-B cells originated this carcinoma, since cellular similarities were identified between these lymphocyte progenitors and MCC, such as the terminal deoxynucleotidyl transferase (TdT) enzyme and the PAX5 gene.12 More recently, through advances in research, theories of other probable origins have gained notoriety. Initially, the influence of epigenetics on the development of MCC was shown to be relevant. It has been postulated that the hypermethylation of genes that are important in cell cycle control can silence them, and the resulting cell divisions favor the emergence of MCC.13 Furthermore, in a case involving MCC cells and squamous cell carcinoma, tests identified mutational similarities between them. These results revived discussion that epithelial cells could also be the cellular origin of MCC.14 Thus, due to extensive discussion about the cell responsible for MCC (Figure 2), we conducted this review gather and examine the existing evidence.
MCC, a rare and aggressive type of skin cancer, is highly lethal, highly metastatic, and has neuroendocrine characteristics. This tumor presents as a painless, fast-growing, smooth-surfaced, firm, erythematous-violet skin lesion (Figure 1).15-17 The main risk factors for MCC can be summarized in the acronym AEIOU: A – asymptomatic; E – expanding rapidly; I – immune suppression; O – older than 50 years of age; U – ultraviolet-exposed site.18 Furthermore, it is common for MCC to appear in individuals with a history of skin cancer, such as squamous cell carcinoma or basal cell carcinoma of the skin.19 The highest incidence of lesions appear in areas with greater exposure to solar radiation, such as the head, neck, chest, and arms. MCC is staged according to the TNM system, i.e., the extent of the primary tumor (T - tumor), the involvement of regional lymph nodes (N - nodes) and the presence of distant metastases from the regional lymph nodes (M - metastasis).20 According to recent American Joint Committee on Cancer criteria, stages I and II refer to tumors in the skin, stage III refers to disease in regional lymph nodes, and stage IV refers to metastatic disease. Stage 0 indicates a small localized lesion, stage I indicates a localized lesion up to 2 cm (T1). Stage IIA indicates localized lesion between 2 and 5 cm (T2) or larger than 5 cm (T3) without lymph node involvement. Stage IIB indicates a tumor that has invaded muscle, cartilage, fascia, or bone tissue (T4) without lymph node involvement. Stage IIIA indicates a tumor of any size that has metastasized to regional lymph nodes according to histopathological analysis (sentinel lymph node or in-transit metastasis). Stage IIIB indicates a tumor of any size that has metastasized to regional lymph nodes according to clinical examination. Stage IV indicates metastasis beyond regional lymph nodes.21 MCC is also classified into 3 histological subtypes: trabecular, which is less common and is usually related to mixed tumors; intermediate, which is more common and has a high rate of mitosis; and small cell, which is similar to small cell carcinomas characteristic of other organs and tissues.22 Regarding histopathology, tumor cells are located in the dermis and may affect subcutaneous or deeper tissue. They consist of small, monomorphic cells, circular or oval in shape, with vesicular nuclei and scarce cytoplasm.23
Initially known as trabecular carcinoma of the skin, this type of tumor appears most commonly in the dermis and subcutaneous tissue. The current name derives from the high similarity between tumor cells and MCs, which are light touch mechanoreceptors located around the base of hair follicles. The lesions present neuroendocrine markers, which are also present in MCs, such as cytokeratin 20, CD56, neuron-specific enolase NSE, chromogranin A and synaptophysin, in addition to neurofilament expression.24-28 However, despite the similarity between tumor cells and MCs, it is unlikely that this cell lineage is the origin of the lesion, since it arises in the dermis and MCs are located in the epidermis.29,10 There are four main hypotheses about the cells that give rise to MCC: dermal fibroblasts, progenitor cells derived from the embryonic neural crest, epithelial cells, and pre- and pro-B cells. Carcinoma emergence may be related to viral integration of MCPyV genetic material into cellular DNA or to mutations in cellular DNA caused by the action of ultraviolet radiation.30,31 Despite the difference in origin, virus-positive MCCs, in which the presence of viral DNA can be identified, and virus-negative MCCs, in which the presence of viral DNA cannot be identified, present in a similar way. They grow rapidly, are painless, have erythematous coloration or are similar in color to the patient's skin tone, and affect the dermal tissue.32,33 Both have neuroendocrine histology, presenting cytokeratins, such as cytokeratin 20, and neuroendocrine markers, such as synaptophysin, chromogranin A, and INSM1. Virus-negative MCC lesions tend to be more aggressive, since they involve numerous genetic mutations and a greater chance of metastasis and recurrence.34 In most cases, MCC is diagnosed late, since the lesion is painless and may resemble other skin tumors. Biopsy, followed by histopathological analysis, is the main diagnostic tool.
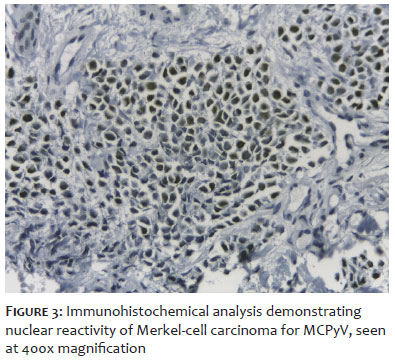
MCPyV, a member of the Polyomaviridae family, is a non-enveloped circular double-stranded DNA virus that is frequently detected in various skin cells. Most people become infected with MCPyV asymptomatically during childhood, so the virus can be found on the skin of most adults.35 Most cases of MCC, a rare and aggressive type of skin tumor, are related to MCPyV infection. The viral genome consists of an early region and a late region, separated by a non-coding control region. The early region expresses genes that encode two T antigens: large T antigen, which is required for viral replication; and small T antigen, which interacts with host cell proteins and amplifies viral replication.4 The late region expresses genes that encode the viral capsid proteins, VP1, VP2, and VP3, which are expressed after DNA replication has initiated and encode a viral microRNA that regulates the expression of early genes.36 MCPyV is responsible for most cases of MCC, with the viral genome integrating into that of tumor cells in most of these lesions, in addition to expressing truncated large T antigen.37 The integration of MCPyV into a person's genome occurs through accidental fragmentation of DNA at random sites during viral replication, without involving the cell's tumor suppressor genes.38 In immunohistochemical analysis, MCPyV can be detected in cells affected by MCC (Figure 3).39 Under normal conditions, large T antigen (among other factors) is responsible for activating the p53 gene, a protein with a tumor suppression function. However, when large T antigen is truncated, the cell proliferation rate increases as a consequence of non-activation of the p53 gene, which can lead to the emergence of a tumor. In addition, it is known that polyomavirus plays an important role in the development of MCC, since it interferes with several cell signaling pathways, although it is not clear which steps are affected.1 It is still not known for sure which host cells maintain latent MCPyV infection.2
MCPyV infects dermal fibroblasts located just beneath the epidermal basement membrane and in close proximity to hair follicles.40 It is possible that the virus infects the dermal cells surrounding hair follicles and uses the follicular space as a means to spread to the skin surface and infect new hosts.2 The migration of infected fibroblasts to injured areas may be another way of transmitting viruses from the cells that store them to deeper layers of the skin, which could lead to the development of carcinoma.2 Although insufficient in itself, MCPyV infection can lead to MCC, and it can be inferred that the cells that originate this tumor, or at least virus-positive MCC, are those susceptible to infection by the virus – the fibroblasts of the dermis close to the hair follicles.41
Neural crest cells emerge during the neurulation process, which occurs around the fourth week of gestation, and are the result of the neural tube. These cells migrate to different areas of the embryo, differentiating into a wide variety of cell types, such as dermal cells.42 Based on this, it was hypothesized that MCs, neuroendocrine cells of the epidermis, originate from the neural crest, since in the fetal dermis these cells are associated with small unmyelinated nerves. In some cases, nerve-associated MCs can be observed crossing the basal layer that separates the dermis and epidermis.43
It is known that MCC may not originate from the cells that give it its name, although there is no certainty about its true cellular origin. It has been hypothesized that embryonic neural crest cells are responsible for this tumor, mainly due to the fact that the pathogenic mechanisms that produce MCC are similar to those of neoplasms with a known origin in neural crest derivatives.11
Tumor development is partly related to the inactivation or loss of tumor suppressor genes. In neoplasms originating from the neural crest, it is common for these genes to be lost through deletions in the short arm of chromosome 1 of the cellular genetic material.44 The tumor cells of most MCCs also present mutations in the distal region of the short arm of chromosome 1, which suggests that this carcinoma originates from the same cell type that gives rise to tumors such as melanoma and neuroblastoma, which are derived from neural crest cells.44 Furthermore, MCC cells express telomerase activity, an enzyme active only in nerve cells, which could indicate that this type of cancer originates from cells with nerve function, such as those that differentiate from embryonic neural crest cells.11
The hypothesis that MCC arises from phenotypic variations in epithelial cells is highly relevant. This theory is based on the possibility of cellular differentiation in epithelial cells, which allows them to acquire neuroendocrine characteristics similar to those of MCs.3 Historically, the principles which indicate that epithelial cells originate MCC have not been very well accepted, with two arguments standing out: first, that MCCs are most frequently found in the dermis and, second, that case reports supporting epithelial origin are rare. It should also be considered that, according to current evidence, MCPyV infection is insufficient to induce the transformation of epithelial cells into neoplastic cells.41 However, recent studies indicate that MCC may originate from alterations in pre-existing epithelial neoplastic cells. In such studies, MCPyV-positive and MCPyV-negative epidermal cells may be precursors to MCC. The theory that gained prominence in these studies is based on clinical reports in which MCC is associated with some other type of epithelial neoplasia. Researchers believe it is possible that clonal diversification of the existing neoplastic population occurs, which acquires the MCC phenotype and, as a result, results in a case of MCC associated with another carcinoma.3 In MCPyV-negative cases, the epithelial origin hypothesis is supported by case studies of MCC associated with squamous cell carcinoma in situ. Based on the genetic sequencing of the cell population, mutational similarities were observed between the two carcinomas, such as TP53 protein and RB1, which are important tumor suppressors. Thus, an etiological relationship between the two carcinomas was suggested, strengthening the theory of epithelial origin in a case of virus-negative MCC.14 However, in MCPyV-positive cases, the influence of viral genetic material and oncogene expression is considered. It is believed that, together, these factors create a cellular environment favorable to the development of MCC from a pre-existing neoplasia.14 A recently published case report suggests that a virus-positive MCC developed from a trichoblastoma. Morphological examination revealed a well-defined tumor with morphological aspects of a trichoblastoma at the edges and, in the center, characteristics of MCC. Furthermore, when quantitative PCR was performed, a significant viral load of MCPyV was observed in the sample. To suggest the genetic relationship between MCC and trichoblastoma, genetic sequencing was performed and mutational similarities were identified between the two neoplasms, suggesting that the integration of MCPyV into trichoblastoma influences the development of MCC.45
The initial stages of lymphoid tissue consist of pro-B and pre-B cells. Until mature B lymphocytes are formed, the progenitor cells mature, passing through the pro-B and pre-B stages, respectively.46 In the pro-B stage, the cells express markers such as CD19, CD20, and CD40, whereas in the pre-B stage, the cell expresses the markers TdT, CD79, and CD10, in addition to initiating the formation of pre-BCR, an immature B cell receptor.47 Evidence supporting the origin of MCC from B lymphocyte progenitors is based on cellular similarities between pro-B/pre-B cells and MCC samples. TdT is a DNA polymerase characteristic of T lymphocytes, lymphoma and/or lymphoblastic leukemia, and lymphocyte precursors in the bone marrow. This enzyme contributes to the elongation of nucleotide chains during the early development of B cells. Immunohistochemical studies have identified positive reactivity for TdT in 73% of MCC samples, which suggests that the cellular origin of MCC may be related to B lymphocyte precursors.48 It is also important to consider the PAX5 gene, which plays an important role in the origin of B cells in identifying this cell lineage by inducting CD19 marker expression during the development of the lymphocyte lineage.49 An immunohistochemical study identified the presence of this gene in 89% of MCC samples. Furthermore, additional tests performed on these samples identified TdT in 78% of MCC samples. These findings indicate that pro-B and pre-B cells may be responsible for the origin of MCC.12 Taken together, the MCC cellular expression data reflect biochemical similarities between MCC and B lymphocyte progenitor cells. Thus, there may be an ancestral relationship between pro-B and pre-B cells and MCC, i.e., pre-B and pro-B cells could transform into tumor cells before their maturation process is completed.50
Epigenetics refers to heritable and reversible modifications of the genome that can control gene expression without altering the DNA sequence. This control can occur through post-translational modifications of histone proteins, DNA methylation, or microRNA expression, processes that cause instability in cells and can alter gene expression, eventually leading to carcinogenesis.51 DNA methylation involves the addition of a methyl group to the carbon 5 position of a nucleotide, silencing the gene that contains it by inhibiting transcription or recruiting corepressor complexes for chromatin remodeling. Methylation events, mainly in promoter regions of tumor suppressor or tumor-related genes, have already been demonstrated in the progression of some types of cancer, such as melanoma, allowing not only the identification of clinical useful epigenetic biomarkers, but the elucidation of their tumorigenesis as well.52 Some hypermethylated genes have already been described in MCC, such as CpG islands in the RASSF1A promoter and in CDKN2A (p14ARF).13 RASSF1A is a tumor suppressor gene that participates in the regulation of the cell cycle and apoptosis.53 p14ARF is a protein encoded by the CDKN2A gene, capable of blocking the cell cycle in the G1 and G2 phases and inhibiting the growth of cancer cells by indirectly activating the p53 tumor suppressor gene.54 Thus, the silencing of these genes observed in MCC suggests that methylation changes may be directly involved in carcinogenesis.13 Furthermore, although MCPyV infection is found in approximately 80% of MCC cases, which may contribute to pathogenesis, there is evidence that viral infection is unrelated to epigenetic inactivation, which indicates that these events are independent.30,53 In MCC patient samples, one tested cell line, MCC13, had greater correspondence with the methylation pattern of small-cell lung cancer than the pattern found in MCC. Since the methylation profiles of these tumor types are unique, the results could indicate that MCC involves cells derived from metastatic cells of another type of carcinoma with similar clinical and pathological characteristics.13 DNA methylation can also be used as an indicator of an individual's biological age.55 Therefore, the development of clocks to estimate epigenetic age by measuring the cumulative effects of methylation over the years facilitates research into issues such as cancer and aging.56 A low DNA methylation age is found in stem cells, and the induction of pluripotency is associated with cell rejuvenation.57 Furthermore, an accelerated epigenetic age reflects the difference between DNA methylation age and chronological age.55 A study revealed that the DNA methylation age of MCC samples was significantly lower than the patients' chronological age, regardless of the presence of MCPyV. Low DNA methylation age could indicate the pluripotency of these cells, a hypothesis corroborated by the idea of trilinear MCC differentiation, since the concomitant expression of epithelial, neuroendocrine, and pre- and pro-B lymphocyte cell lineages suggests that stem cells could be the origin of this tumor. However, the results were negative when MCC samples underwent pluripotency analysis, revealing a paradoxical situation of epigenetic youth without pluripotency57 (Table 2).
The origin of MCC, first described in 1972, has not yet been clearly defined. So far, only hypotheses have been postulated to explain its development. The oldest hypothesis is that the cells are derived from the embryonic neural crest, which is based on similarities between the carcinogenesis of MCC and tumors that clearly originate from the neural crest, such as telomerase activity and the loss of tumor suppressor genes at similar gene loci.11,44 Despite the importance of this similarity, this appears to be the least likely hypothesis at present, since other similarities are missing between MCC cells and melanoma and neuroblastoma cells, tumors originating in the embryonic neural crest. Furthermore, it is possible that MCC originates from pre-B and pro-B cells, given the similarities between these cells and tumor cells of the cancer in question. This similarity is due to the presence of TdT in MCC samples, an enzyme characteristic of lymphocytes in early development.48 The presence of CD19 in carcinoma cells also supports this hypothesis, since this marker is encoded by the PAX5 gene, which is characteristic of lymphocyte lineage cells that are also present in the tumor.49 When viral genetic material can be detected in tumors, the cells most likely to become neoplastic are the dermal fibroblasts, which are located close to the hair follicles, since these cells are susceptible to viral infection.2 Upon infection, the polyomavirus' genetic material is integrated into the cellular DNA, increasing the rate of cell proliferation, in addition to interfering in important cell signaling pathways, which can lead to the development of cancer.1,38 It is also possible that MCC originates from pre-existing neoplastic cells that develop from epithelial cells. This hypothesis is based on the presence of mutations in the TP53 and Rb1 genes, which are tumor suppressors in both MCC and squamous cell carcinoma in the absence of MCPyV infection.14 In virus-positive tumors, the expression of viral genetic material concomitantly with the expression of oncogenes may favor carcinogenesis, a theory reinforced by samples containing similar morphological and genetic characteristics between MCC and other skin tumors, such as trichoblastoma. Furthermore, cell line analysis indicates that the methylation pattern of MCC corresponds to other tumor types, such as small cell lung cancer, showing that MCC may originate from diverse metastatic cells and is not restricted to epithelial cells. Epigenetics is an important factor in elucidating tumor development. The genome modification hypothesis is supported by the observation of tumor suppressor genes silenced by hypermethylation mechanisms in MCC samples.13 Furthermore, epigenetic analysis has shown that it is unlikely that stem cells are the origin of MCC, since samples have tested negative for pluripotency. Based on the data, there is still not enough information to determine the exact origin of MCC. There is a high probability that this neoplasm has more than one cellular origin and may be influenced by mechanisms of epigenetic modification. We believe that the cells most likely to originate MCC are poorly differentiated cells that can be specified in other cell types, which might explain a multiple origin for this carcinoma. Nevertheless, studies such as Chteinberg et al. have found negative pluripotency results for MCC, which prevents an objective definition of the origin of MCC until further research can be conducted.57
Fernanda Guimarães Souza
ORCID: 0009-0004-8385-0154
Formal analysis; approval of the final version of the manuscript; conceptualization; writing – original draft; data curation; intellectual participation in propaedeutic and/or therapeutic conduct of the studied cases; critical review of the literature; writing – review & editing
Daniela Pessanha dos Santos
ORCID: 0009-0002-1490-9613
Formal analysis; approval of the final version of the manuscript; conceptualization; writing – original draft; obtaining, data curation; intellectual participation in the propaedeutic and/or therapeutic conduct of the studied cases; critical review of the literature; writing – review & editing.
Luiza Oliveira Ribeiro
ORCID: 0009-0009-7355-9378
Formal analysis; approval of the final version of the manuscript; conceptualization; writing – original draft; obtaining, data curation; intellectual participation in the propaedeutic and/or therapeutic conduct of the studied cases; critical review of the literature; writing – review & editing.
Thiago Rubim Batista Bellot Nascimento
ORCID: 0000-0003-3909-5935
Approval of the final version of the manuscript; conceptualization; data curation; formal analysis; supervision; intellectual participation in propaedeutic and/or therapeutic conduct of the studied cases; critical review of the literature; writing – review & editing.
Flávio Barbosa Luz
ORCID: 0000-0001-5454-8950
Approval of the final version of the manuscript; conceptualization; data curation; formal analysis; supervision; intellectual participation in propaedeutic and/or therapeutic conduct of the studied cases; critical review of the literature; writing – review & editing.
1. Uchi H. Merkell cell carcinoma: an update and immunotherapy. Front Ocol. 2018;8:48.
2. Krump NA, You J. From Merkel cell polyomavirus infection to Merkel cell carcinoma oncogenesis. Front Microbiol. 2021;12:739695.
3. Yang JF, You J. Merkel cell polyomavirus and associated Merkel cell carcinoma. Tumour Virus Res. 2022;13:200232.
4. Pietropaolo V, Prezioso C, Moens U. Merkel cell polyomavirus and Merkel cell carcinoma. Cancers (Basel). 2020;12(7):1774.
5. Toker C. Trabular carcinoma of the skin. Arch Dermatol.1972;105(1):107-110.
6. Bataille A, Le Gall C, Misery L, Talagas M. Merkel cells are multimodal sensory cells: a review of study methods. Cells. 2022;11(23):3827.
7. Tang CK, Toker C. Trabecular carcinoma of the skin: an ultrastructural study. Cancer. 1978;42(5):2311-2321.
8. Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. I. A clinopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9(2): 95-108.
9. Sunshine JC, Jahchan NS, Sage J, Choi J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene. 2018;37(11):1409-1416.
10. Tilling T, Moll I. Which are the cells of origin in Merkel cell carcinoma? J Skin Cancer. 2012;2012:680410.
11. Suárez C, Rodrigo JP, Ferlito A, Devaney KO, Rinaldo A. Merkel cell carcinoma of the head and neck. Oral Oncology. 2004; 40(8):773-779.
12. Kolhe R, Reid MD, Lee JR, Cohen C, Ramalingam P. Immunohistochemical expression of PAX5 and TdT by Merkel cell carcinoma and pulmonary small cell carcinoma: a potential diagnostic pitfall but useful discriminatory marker. Int J Clin Exp Pathol. 2013. 6(2):142-147.
13. Gujar H, Mehta A, Li HT, Tsai YC, Qiu X, Weisenberger DJ, et al. Characterizing DNA methylation signatures and their potential functional roles in Merkel cell carcinoma. Genome Med. 2021; 13(1):130.
14. Thibault K. Evidence of an epithelial origin of Merkel cell carcinoma. Mod Pathol. 2022;35(4):446-448.
15. Swann MH, Yoon J. Merkel cell carcinoma. Semin Oncol. 2007;34(1):51-56.
16. Hitchcock CL, Bland KI, Laney III RG, Franzini D, Harris B, Copeland EM. Neuroendocrine (Merkel cell) carcinoma of the skin. Its natural history, diagnosis, and treatment. Ann Surg. 1988;207(2):201-207.
17. Pectasides D, Pectasides M, Economopoulos T. Merkel cell cancer of the skin. Ann Oncol. 2006; 17(10):1489-1495.
18. Walsh NM, Cerroni L. Merkel cell carcinoma: a review. J Cutan Pathol. 2021;48(3):411-421.
19. Cerroni L, Kerl H. Primary cutaneous neuroendocrine ( Merkel cell) carcinoma in association with squamous – and basal – cell carcinoma. Am J Dermatopathol. 1997;19(6):610-613.
20. 20. Cornejo C, Miller CJ. Merkel cell carcinoma: updates on staging and management. Dermatol Clin. 2019;37(3):269-277.
21. Harms KL, Healy MA, Nghiem P, Sober AJ, Johnson TM, Bichakjian CK, et al. Analysis of prognostic factors from 9387 Merkel cell carcinoma cases forms the basis for the new 8th edition AJCC staging system. Ann Surg Oncol. 2016;23(11):3564-3571.
22. Baba PUF, Rasool Z, Younas Khan I, Cockerell CJ, Wang R, Kassir M, et al. Merkel cell carcinoma: from pathobiology to clinical management. Biology (Basel). 2021;10(12):1293.
23. Fried I, Cerroni L. [Merkel cell carcinoma]. Patholege. 2014;35(5):467-475.
24. Moll I, Kuhn C, Moll R. Cytokeratin 20 is a general marker of cutaneous Merkel cells while certain neuronal proteins are absent. J Invest Dermatol. 1995;104(6):910-915.
25. Eispert AC, Fuchs F, Brandner JM, Houdek P, Wladykowski E, Moll I. Evidence for distinct populations of human Merkel cells. Histochem Cell Biol. 2009;132(1):83-93.
26. Kurokawa M, Nabeshima K, Akiyama Y, Maeda S, Nishida T, Nakayama F, et al. CD56: a useful marker for diagnosing Merkel cell carcinoma. J Dermatol Sci. 2003;31(3):219-224.
27. Llombart B, Monteagudo C, Lopez-Guerrero JA, Carda C, Jorda E, Sanmartin O, et al. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology. 2005;46(6):622-634.
28. Shah IA, Netto D, Schlageter MO, Muth C, Fox I, Manne RK. Neurofilament immunoreactivity in Merkel-cell tumors: a differentiating feature from small-cell carcinoma. Mod Pathol. 1993;6(1):3-9.
29. Moll I, Roessler M, Brandner JM, Eispert AC, Houdek P, Moll R. Human Merkel cells – aspects of cell biology, distribution and functions. Eur J Cell Biol. 2005;84(2-3):259-271.
30. Feng H, Shuda M, Chang Y, Moore PS. Clonal Integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096-1100.
31. Wong SQ, Waldeck K, Vergara IA, Schröder J, Madore J, Wilmott JS, et al. UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. 2015;75(24):5228-5234.
32. DeCaprio JA. Molecular pathogenesis of Merkel cell carcinoma. Annu Rev Pathol. 2021;16:69-91.
33. Ahmed MM, Cushman CH, DeCaprio JA. Merkel cell polyomavirus: oncogenesis in a stable genome. Viruses. 2021;14(1):58.
34. Jaeger T, Ring J, Andres C. Histological, immunohistological, and clinical features of Merkel cell carcinoma in correlation to Merkel cell Polyomavirus status. J Skin Cancer. 2012;2012:983421.
35. Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5(3):e1000363.
36. Gjoerup O, Chang Y. Update on human polyomaviruses and cancer. Adv Cancer Res. 2010;106:1-51.
37. Csoboz B, Rasheed K, Sveinbjørnsson B, Moens U. Merkel cell polyomavirus and non‐Merkel cell carcinomas: guilty or circumstantial evidence? APMIS. 2020;128(2):104-120.
38. Starrett GJ, Marcelus C, Cantalupo PG, Katz JP, Cheng J, Akagi K, et al. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. mBio. 2017;8(1):e02079-16.
39. Nascimento TRBB. Detecção de poliomavírus de células de Merkel em carcinomas de queratinócitos pela imuno-histoquímica [dissertação]. [Niterói]: Universidade Federal Fluminense; 2023. 95 p.
40. Liu W, Yang R, Payne AS, Schowalter RM, Spurgeon ME, Lambert PF, et al. Identifying the target cells and mechanisms of Merkel cell polyomavirus infection. Cell Host Microbe. 2016;19(6):775-787.
41. Harms PW, Harms KL, Moore PS, DeCaprio JA, Nghiem P, Wong MKK, et al. The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nat Rev Clin Oncol. 2018;15(12):763-776.
42. Schoenwolf GC, Larsen WJ, Bleyl SB, Brauer PR, Francis-West PH. Larsen's Human Embryology. Churchill Livingstone.; 2009.
43. Munde P, Khandekar S, Dive A, Sharma A. Pathophysiology of Merkel cell. J Oral Maxillofac Pathol. 2013;17(3):408-412.
44. Harnett PR, Kearsley JH, Hayward NK, Dracopoli NC, Kefford RF. Loss of allelic heterozygosity on distal chromosome 1p in Merkel cell carcinoma. A marker of neural crest origins? Cancer Genet Cytogenet. 1991;54(1):109-113.
45. Kervarrec T, Aljundi M, Appenzeller S, Samimi M, Maubec E, Cribier B, et al. Polyomavirus-positive Merkel cell carcinoma derived from a Trichoblastoma suggests an epithelial origin of this Merkel cell carcinoma. J Invest Dermatol. 2020;140(5):976-985.
46. Urbanczyk S, Stein M, Schuh W, Jäck HM, Mougiakakos D, Mielenz D. Regulation of energy metabolism during early B lymphocyte development. Int J Mol Sci. 2018;19(8):2192.
47. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570-1580.
48. Buresh CJ, Oliai BR, Miller RT. Reactivity with TdT in Merkel cell carcinoma: a potencial diagnostic pitfall. Am J Clin Pathol. 2008;129(6):894-898.
49. Patton DT, Plumb AW, Abraham N. The survival and differentiation of pro- B and pre-B cells in the bone marrow is dependent on IL-7Rα Tyr449. J Immunol. 2014;193(7):3446-3455.
50. zur Hausen A, Rennspiess D, Winnepenninckx V, Speel EJ, Kurz AK. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry. Cancer Res. 2013;73(16):4982-4987.
51. Rotondo JC, Mazziotta C, Lanzillotti C, Tognon M, Martini F. Epigenetic dysregulations in Merkel cell polyomavirus-driven Merkel cell carcinoma. Int J Mol Sci. 2021;22(21):11464.
52. Greenberg ES, Chong KK, Huynh KT, Tanaka R, Hoon DSB. Epigenetic biomarkers in skin cancer. Cancer Lett. 2014;342(20):170-177.
53. Helmbold P, Lahtz C, Enk A, Herrmann-Trost P, Marsch WCh, Kutzner H, et al. Frequent occurrence of RASSF1A promoter hypermethylation and Merkel cell polyomavirus in Merkel cell carcinoma. Mol Carcinog. 2009;48(10):903-909.
54. Sunwoo HH, Suresh MR. The immunoassay handbook: theory and applications of ligand binding, ELISA and related techniques [Internet]. 4th ed. Elsevier; 2013. Part 9, Chapter 12, Cancers Markers; p 833-856.
55. Noroozi R, Ghafouri-Fard S, Pisarek A, Rudnicka J, Spólnicka M, Branicki W, et al. DNA methylation-based age blocks: from age prediction to age reversion. Ageing Res Rev. 2021;68:101314.
56. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115.
57. Chteinberg E, Vogt J, Kolarova J, Bormann F, Van Den Oord J, Speel EJ, et al. The curious case of Merkel cell carcinoma: epigenetic youth and lack of pluripotency. Epigenetics. 2020;15(12):1319-1324.
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}