


Fernanda Guimarães Souza1; Daniela Pessanha dos Santos1; Luiza Oliveira Ribeiro1; Thiago Rubim Batista Bellot Nascimento2; Flávio Barbosa Luz3
Fonte de financiamento: Nenhuma.
Conflito de interesses: Nenhum.
Data de Submissão: 26/02/2024
Decisão final: 21/11/2024
How to cite this article: Souza FG, Santos DP, Ribeiro LO, Nascimento TRBB, Luz FB. Ontogenisis of Merkel-cell carcinoma. Surg Cosmet Dermatol. 2024;16:e202040354.
Foi realizada uma revisão narrativa para elucidar a ontogênese do carcinoma de células de Merkel. Entre as hipóteses consideradas, há as células derivadas da crista neural embrionária; células-tronco da pele; células epiteliais; células pré e pró-B; fibroblastos da derme; ou células metastáticas de outros tipos de carcinomas. A influência da epigenética também foi considerada. Metodologicamente, para a elaboração do resumo foram selecionados artigos das bases de dados PubMed e SciELO e algumas de suas referências. Os resultados sugerem que é mais provável que o carcinoma de células de Merkel tenha origem múltipla, possivelmente a partir de células pouco diferenciadas.
Keywords: Linhagem Celular Tumoral; Carcinoma de Célula de Merkel; Oncologia; Neoplasias.
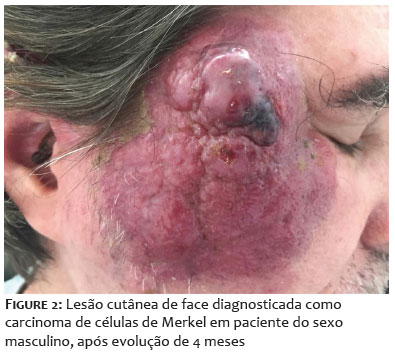
O carcinoma de células de Merkel (CCM), antigamente conhecido como carcinoma trabecular da pele, é um carcinoma raro e agressivo, de característica neuroendócrinas, com alta letalidade e frequência de metástase. Apresenta-se como lesão cutânea firme, de superfície lisa, coloração eritemato-violácea, de rápido crescimento e indolor (Figura 1).1 O tumor é desenvolvido primeiramente na derme, com frequente progressão para o tecido subcutâneo, gerando metástase pelo sistema linfático, características que explicam a sua rápida disseminação, destacando a importância do diagnóstico precoce. Os principais fatores de risco para o desenvolvimento desse tipo de câncer são: idade avançada, pele clara, exposição à radiação ultravioleta (UV) e comprometimento do sistema imunológico. Além disso, sabe-se que aproximadamente 80% dos casos de CCM estão associados ao poliomavírus das células de Merkel (MCPyV), um vírus não encapsulado de DNA circular, pertencente à família Polyomaviridae, que comumente infecta queratinócitos da pele. A maior parte da população tem o primeiro contato com o MCPyV durante a primeira infância, e a maioria das infecções ocorre de forma assintomática, com a permanência do vírus em estado de latência durante toda a vida. Posteriormente, quando mal controlado pelos mecanismos do sistema imunológico, pode originar o CCM (CCM vírus-positivo ou CCMVP). O restante dos casos de CCM não relacionados ao poliomavírus (CCM vírus-negativo ou CCMVN) é causado principalmente por mutações decorrentes da exposição crônica à radiação UV nas células de origem do tumor.2 As lesões compartilham marcadores neuroendócrinos com as células de Merkel (CMs), como a citoqueratina 20 (CK20) e a enolase neurônio-específica (NSE), o que levou ao seu nome.2 Contudo, apesar dessa aparente relação, a linhagem celular que origina esse carcinoma ainda é uma incógnita. As CMs são células pós-mitóticas e, por isso, apresentam potencial oncogênico limitado, além de estarem localizadas na epiderme, fatores que enfraquecem a hipótese de que o CCM se origine a partir desse tipo celular. Ainda, testes indicam que as CMs não respondem efetivamente aos estímulos oncogênicos, inclusive aos antígenos do poliomavírus.3 Foram propostas algumas linhagens celulares que podem originar o CCM: células derivadas da crista neural embrionária; células progenitoras ou células-tronco da pele; células epiteliais; células pré e pró-B; fibroblastos da derme próximos aos folículos pilosos; ou células metastáticas de outros tipos de carcinomas.2-4 Alguns estudos propõem, ainda, que os CCMVNs sejam originados por linhagens celulares diferentes, que convergem para um mesmo fenótipo por meio de reprogramação epigenética.2 As pesquisas voltadas para a definição da oncogênese do CCM são de extrema importância para o estabelecimento de biomarcadores específicos desse câncer. Com isso, serão possíveis avanços médicos que visem ao desenvolvimento de formas mais adequadas para o manejo do CCM, como a elaboração de medidas profiláticas que reduzam a incidência da doença e que melhorem o prognóstico desses pacientes, diminuindo as taxas de metástase e de recidiva, assim como o número de óbitos.
Foi realizada revisão narrativa de literatura por três autores, com base em 57 artigos. As pesquisas iniciais foram realizadas a partir das bases de dados PubMed e SciELO, em línguas inglesa e portuguesa, publicados no período de 2008 a 2022. Os termos utilizados foram: "Merkel Cell Carcinoma" e "Origin". Dos 74 artigos, foram selecionados 42 que abordavam as bases teóricas das principais hipóteses ontológicas do CCM. Adicionalmente, 15 artigos que constavam das referências utilizadas também foram considerados na pesquisa, totalizando 57 artigos. Por fim, com base nesse material científico selecionado, foi desenvolvida a revisão narrativa sobre as diferentes possíveis origens celulares do CCM.
O CCM foi inicialmente descrito em 1972 como carcinoma trabecular da pele e, naquele período, acreditava-se que sua origem era écrina.5 O nome "carcinoma de células de Merkel" foi determinado apenas 6 anos depois, em 1978, após a identificação, pela imuno-histoquímica, da possível relação entre esse carcinoma e as CMs presentes na epiderme, evidenciada na Tabela 1. As CMs são células sensoriais epidérmicas que interagem com neurônios aferentes do mecanorreceptor de baixo limiar Aβ tipo 1 e, por isso, são consideradas importantes na sensação de toque leve.6 A hipótese que relacionava a neoplasia recém-descoberta às CMs justificava-se pelas semelhanças fenotípicas entre elas. Estudos de microscopia eletrônica realizados com amostras de carcinomas trabeculares da pele identificaram a presença de grânulos neurossecretores, o que reforçou a teoria de que a possível origem dessa neoplasia poderia ser as CMs.7 Todavia, mais tarde, alguns estudos encontraram diversos indícios de que as CMs eram uma origem pouco provável deste carcinoma. Entre as evidências que sustentam essa nova perspectiva, destaca-se, primeiramente, a diferença topográfica entre os CCMs e as CMs: as CMs estão presentes na epiderme, camada mais superficial da pele, enquanto os CCMs são majoritariamente encontrados na derme.8 Além disso, ainda que ambos os conjuntos celulares expressem citoqueratinas específicas semelhantes, como a CK20, análises histológicas indicam que a disposição dessas moléculas ocorre de forma distinta em cada tipo celular. Nas CMs, a CK20 é frouxamente organizada, enquanto nos CCMs elas se organizam em espiral ou em arranjos análogos à placa.9 Deve-se considerar, ainda, dados que indicam que os CCMs expressam marcadores não identificados nas CMs, como o receptor tirosina-quinase c-kit e a molécula de adesão CD171.10 Assim, novamente o questionamento a respeito de qual é o tipo celular responsável por originar o CCM veio à tona. As células da crista neural foram inicialmente cotadas, já que o mecanismo patogênico de formação do CCM é similar ao mecanismo de outras neoplasias que possuem origem nessas células.11 Além dessa hipótese, surgiu a teoria de que as células pré-b e pró-b compunham a gênese desse carcinoma, uma vez que foram identificadas similaridades celulares entre essas progenitoras de linfócitos e o CCM, como a enzima desoxinucleotidil transferase terminal (TdT) e o gene PAX5.12 Mais recentemente, por meio dos avanços das pesquisas, teorias que abordam outras prováveis origens adquiriram notoriedade. A princípio, a influência da epigenética no desenvolvimento do CCM mostrou-se relevante. Postula-se que a hipermetilação de genes importantes no controle do ciclo celular é capaz de silenciá-los e, com isso, permitir divisões celulares sucessivas, favorecendo o surgimento do CCM.13 Ademais, foram feitos testes que identificaram similaridades mutacionais entre as células do CCM e as células do carcinoma de células escamosas in situ em um caso que possuía essas neoplasias associadas. Os resultados retomaram a discussão que, até então, tinha pouca notoriedade, sobre a possibilidade de a origem celular do CCM ser também a partir de células epiteliais.14 Desse modo, é perceptível que, ao longo da história (Figura 2), a célula responsável por originar o CCM foi pauta de extensas discussões no âmbito científico. Cabe a nós, portanto, reunir nesta revisão conteúdos que embasam evidências encontradas até hoje.
O CCM é um tipo raro e agressivo de câncer de pele, de alta letalidade, altamente metastático e com características neuroendócrinas. Esse tumor se apresenta como uma lesão cutânea firme, de superfície lisa, coloração eritêmato-violácea, de rápido crescimento e indolor (Figura 1).15-17 Os principais fatores de risco para o desenvolvimento do CCM podem ser resumidos no acrônimo AEIOU: A – asymptomatic ou assintomático; E – expanding rapidly ou expansão rápida; I – immune suppression ou imunossupressão; O – older than 50 years ou idade avançada, acima de 50 anos; U – UV-exposed site ou localização fotoexposta.18 Além disso, é comum o aparecimento desse tumor em indivíduos com história de outra forma de câncer de pele, como o carcinoma de células escamosas ou o carcinoma de células basais da pele.19 As regiões corporais de maior incidência da lesão são cabeça e pescoço, peito e membros superiores, que ficam mais expostas à radiação solar. O CCM é estadiado de acordo com o sistema de estadiamento TNM, conforme a extensão do tumor primário (T – tumor), o acometimento de linfonodos regionais (N – nodes) e a presença de metástases à distância dos linfonodos regionais (M – metastasis).20 Pelos critérios recentemente atualizados pela American Joint Committee on Cancer, os estágios I e II se referem a tumores localizados na pele, III, doença em linfonodos regionais, e IV, doença metastática: estágio 0, lesão localizada de pequena dimensão; estágio I, lesão localizada com até 2 cm (T1); estágio IIA, lesão localizada entre 2 e 5 cm (T2) ou maior que 5 cm (T3), sem acometimento linfonodal; estágio IIB, tumor com invasão de músculo, cartilagem, fáscia ou osso (T4), sem acometimento linfonodal; estágio IIIA, tumor de qualquer tamanho com metástase para linfonodos regionais identificada por meio de análise histopatológica (pesquisa de linfonodo sentinela ou metástases em trânsito); estágio IIIB, tumor de qualquer tamanho, com metástase para linfonodos regionais identificada através do exame clínico; estádio IV, metástase para além dos linfonodos regionais.21 Além disso, o CCM é classificado em 3 subtipos histológicos: trabecular, menos comum e geralmente relacionado a tumores mistos; intermediário, mais comum, com uma alta taxa de mitose; e de pequenas células, similar a carcinomas de pequenas células característicos de outros órgãos e tecidos.22 Quanto à histopatologia, as células tumorais se localizam na derme, podendo acometer tecido subcutâneo ou de maior profundidade, são compostos por células pequenas, monomórficas, de formato circular ou ovalado, com núcleos vesiculares e citoplasma escasso.23
Inicialmente conhecido como carcinoma trabecular da pele, esse tipo de tumor aparece mais comumente na derme e tecido subcutâneo. O nome atual deriva da alta semelhança que as células tumorais têm com as CMs, que são mecanorreceptores para tato leve localizados em torno da base de folículos pilosos. As lesões apresentam marcadores neuroendócrinos, presentes também nas CMs, como a CK20, CD56, enolase neurônio específica NSE, cromogranina A e a sinaptofisina, além da expressão de neurofilamentos.24-28 Contudo, apesar da similaridade das células tumorais com as CMs, é improvável que essa linhagem celular seja a que origina a lesão, uma vez que surge na derme, e as CMs se localizam na epiderme.29,10 Existem quatro hipóteses principais para as células que originam o CCM: fibroblastos da derme, células progenitoras derivadas da crista neural embrionária, células epiteliais e células pré e pró-B. O surgimento do carcinoma pode estar relacionado com a integração viral do material genético do MCPyV ao DNA celular ou com mutações no DNA celular causadas pela ação da radiação UV.30,31 Apesar da diferença de origem, os CCMVPs, nos quais é possível identificar a presença de DNA viral, e os CCMVNs, nos quais não é possível identificar a presença de DNA viral, apresentam-se de forma semelhante. Eles possuem rápido crescimento, ausência de dor, coloração eritematosa ou de cor semelhante ao tom de pele do indivíduo, e ao tecido acometido, a derme.32,33 Ambos têm histologia neuroendócrina, apresentando citoqueratinas, como a CK20, e marcadores neuroendócrinos, como a sinaptofisina, a cromogramina A e a INSM1. As lesões de CCMVN tendem a ser mais agressivas, pois apresentam muitas mutações no material genético celular, além de apresentarem maior chance de metástase e de recidiva.34 O diagnóstico do CCM, em grande parte dos casos, acontece de forma tardia, uma vez que a lesão é indolor e pode se assemelhar a outros tumores cutâneos. A biópsia da lesão, seguida de análise histopatológica, é a principal ferramenta diagnóstica atual.
O MCPyV, integrante da família Polyomaviridae, é um vírus não envelopado de DNA de fita dupla circular, detectado com frequência em diversas células cutâneas. A maior parte das pessoas se infecta com o MCPyV, de forma assintomática, durante a infância, de modo que o vírus pode ser encontrado na pele da maioria dos adultos.35 O desenvolvimento do CCM, um tipo raro e agressivo de tumor cutâneo, na maioria dos casos está relacionado à infecção pelo MCPyV. O genoma viral é composto por uma região precoce e uma região tardia, separadas por uma região de controle não codificadora. A região inicial expressa genes que codificam dois antígenos T: antígeno T maior (LT-Ag), necessário para replicação viral; e antígeno T menor (sT-Ag), responsável pela interação com proteínas da célula hospedeira, além de ampliar a replicação viral.4 A região tardia expressa genes que codificam proteínas do capsídeo viral (VP), VP1, VP2 e VP3, expressas depois que a replicação do DNA foi iniciada e que codificam um microRNA viral que regula a expressão dos genes precoces.36 O MCPyV é responsável pela maior parte dos CCMs, havendo a integração do genoma viral ao das células tumorais na maior parte dessas lesões, além da expressão do LT-Ag truncado.37 A integração do MCPyV ao genoma do indivíduo ocorre por uma fragmentação acidental do DNA durante a replicação viral, em sítios aleatórios, sem envolvimento de genes supressores tumorais da célula.38 Na análise imuno-histoquímica, é possível detectar a presença do MCPyV em células acometidas pelo CCM (Figura 3).39 Em normalidade, o LT-Ag é responsável, entre outros fatores, pela ativação do gene p53, uma proteína com função de supressão tumoral. Contudo, quando ocorre o truncamento do LT-Ag, há um aumento na taxa de proliferação celular, consequência da não ativação do gene p53, de maneira que pode haver surgimento de tumor. Além disso, sabe-se que o poliomavírus tem importante papel no desenvolvimento do CCM, pois interfere em diversas vias de sinalização das células, apesar de não haver clareza sobre quais etapas são afetadas.1 Ainda não se sabe ao certo quais são as células hospedeiras que mantêm a infecção latente do MCPyV.2
O MCPyV infecta fibroblastos da derme localizados logo abaixo da membrana basal da epiderme e nas proximidades de folículos pilosos.40 É possível que o vírus infecte as células da derme que cercam o folículo piloso e use o espaço folicular como meio para se disseminar para a superfície da pele e infectar novos hospedeiros.2 A migração de fibroblastos infectados pelo vírus para locais lesionados pode ser outra forma de transmissão dos vírus das células que os armazenam para camadas mais profundas da pele, podendo ocasionar o desenvolvimento do carcinoma.2 Tendo em vista que a infecção pelo MCPyV pode acarretar o surgimento do CCM, apesar de não ser suficiente para tal, é possível inferir que as células que originam esse tumor, ou, pelo menos, os CCMVP, sejam as células susceptíveis à infecção pelo vírus – os fibroblastos da derme próximos aos folículos pilosos.41
As células da crista neural surgem durante o processo de neurulação, que ocorre por volta da 4ª semana de gestação, e são frutos do tubo neural. Essas células migram para diferentes áreas do embrião, diferenciando-se em uma grande variedade de tipos celulares, como, por exemplo, células da derme.42 Com base nisso, foi criada a hipótese de que as CMs, células neuroendócrinas da epiderme, originam-se da crista neural, uma vez que na derme fetal essas células estão associadas com pequenos nervos amielínicos. Em alguns casos, CMs associadas a nervos podem ser identificadas cruzando a camada basal que separa derme e epiderme.43
Sabe-se que o CCM pode não ter origem a partir das células que lhe dão nome, apesar de não haver certeza sobre sua verdadeira origem celular. Uma das hipóteses postuladas sobre o tipo celular que origina esse tumor é baseada nas células da crista neural embrionária. Tal hipótese decorre, principalmente, do fato de que os mecanismos patogênicos que dão origem ao CCM são semelhantes aos mecanismos patogênicos de neoplasias com origem conhecida a partir de derivados da crista neural.11
O desenvolvimento de tumores está, em parte, relacionado com a inativação ou com a perda de genes supressores de tumor. Em neoplasias que se originam da crista neural, é comum que esses genes sejam perdidos a partir de deleções no braço curto do cromossomo 1 do material genético celular.44 As células tumorais de grande parte dos CCMs apresentam, também, mutações na região distal no braço curto do cromossomo 1, fato que sugere que esse carcinoma tenha origem a partir do mesmo tipo celular que origina tumores como o melanoma e o neuroblastoma – derivados das células da crista neural.44 Além disso, as células do CCM expressam atividade da telomerase, enzima ativa apenas em células nervosas, o que pode indicar que esse tipo de câncer se origine de células com função nervosa, como as que se diferenciam a partir das células da crista neural embrionária.11
A hipótese de que o CCM advém de variações fenotípicas de uma célula epitelial é altamente relevante. Essa teoria é pautada na possibilidade de ocorrer um processo de diferenciação celular das células epiteliais, que as permite adquirir características neuroendócrinas similares às das CMs.3 Historicamente, os princípios que apontam as células epiteliais como células de origem do CCM não são muito bem aceitos, com destaque para dois argumentos: que os CCMs são mais frequentemente encontrados na derme e que as notificações de casos que sustentam essa provável origem epitelial são raras. Deve-se considerar, ainda, que as evidências atuais indicam que a infecção pelo MCPyV não é suficiente para induzir a transformação das células epiteliais em células neoplásicas.41 Todavia, estudos recentes indicam a possível origem do CCM a partir de alterações em células neoplásicas epiteliais pré-existentes. Em tais pesquisas, as amostras de células da epiderme MCPyV-positivo e MCPyV-negativo são possíveis precursoras do CCM. A teoria que ganhou destaque nessas pesquisas atuais é pautada em relatos clínicos em que há o CCM associado a algum outro tipo de neoplasia epitelial. Pesquisadores acreditam que é possível que ocorra uma diversificação clonal da população neoplásica já existente, que adquire o fenótipo do CCM e, com isso, resulta em um caso de CCM associado a um outro carcinoma.3 A hipótese da origem epitelial em um caso de MCPyV-negativo é sustentada por estudos de um caso de CCM associado ao carcinoma de células escamosas in situ. A partir do sequenciamento genético da população celular, foram constatadas similaridades mutacionais entre os dois carcinomas, como na proteína TP53 e na RB1, que são importantes supressores tumorais. Desse modo, a relação etiológica entre os dois carcinomas foi sugerida, fortalecendo a teoria da origem epitelial em um caso de CCMVN.14 Em um caso positivo para o MCPyV, por sua vez, é considerada a influência da expressão de material genético viral e de oncogenes. Acredita-se que, juntos, esses fatores compõem um ambiente celular favorável ao desenvolvimento do CCM a partir de uma neoplasia pré-existente.14 Um relato de caso recentemente publicado sugere o desenvolvimento de um CCMVP a partir de um tricoblastoma. O exame morfológico constatou um tumor bem delimitado, que nas bordas apresentava aspectos morfológicos de tricoblastoma e, no centro, características de CCM. Além disso, quando realizado PCR quantitativo, foi demonstrada uma relevante carga viral de MCPyV na amostra. Para sugerir a relação genética entre o CCM e o tricoblastoma, foi efetuado um sequenciamento genético e foram identificadas similaridades mutacionais entre as duas neoplasias, sugerindo que a integração do MCPyV ao tricoblastoma tenha influência no desenvolvimento do CCM.45
As células pró-B e pré-B compõem os estágios iniciais do tecido linfoide. Até que haja a formação do linfócito B maduro, há a maturação da célula progenitora, que passa pelos estágios pró-B e pré-B, respectivamente.46 No estágio pró-B, a célula expressa marcadores como CD19, CD20 e CD40, ao passo que no estágio pré-B, a célula expressa os marcadores TdT, CD79 e CD10, além de iniciar a formação do pré-BCR, um receptor imaturo de células B.47 As evidências que sustentam a origem do CCM a partir de progenitores de linfócitos B baseiam-se em similaridades celulares entre as células pró e pré-B e as amostras de CCM. O TdT corresponde a uma DNA polimerase que é característica de linfócitos T, de linfoma e/ou leucemia linfoblástica e de precursores de linfócitos na medula óssea. Essa enzima possui relevância no alongamento das cadeias de nucleotídeos durante o desenvolvimento inicial das células B. Estudos imuno-histoquímicos identificaram reatividade positiva para TdT em 73% das amostras de CCMs utilizadas, o que sugere que a origem celular do CCM pode estar relacionada aos precursores de linfócitos B.48 É relevante considerar, ainda, o gene PAX5, que desempenha uma importante função na origem das células B, atuando como base para a identificação dessa linhagem celular por meio da indução da expressão do marcador CD19 durante o desenvolvimento da linhagem linfocitária.49 Um estudo imuno-histoquímico identificou a presença desse gene em 89% das amostras de CCM. Além disso, testes adicionais realizados nessas amostras identificaram a presença de TdT em 78% dos CCM. Esses achados indicam que as células pró e pré-B podem ser as responsáveis por originar o CCM.12 Em conjunto, os dados a respeito da expressão celular nas amostras de CCM utilizadas nos estudos citados refletem similaridades bioquímicas entre o CCM e as células progenitoras de linfócitos B. Com isso, é possível que exista uma relação de ancestralidade entre as células pró-B e pré-B e o CCM, de modo que o desenvolvimento do CCM ocorra a partir da transformação das células pré-B e pró-B em células tumorais antes que o seu processo de maturação seja concluído.50
A epigenética corresponde a modificações herdáveis e reversíveis do genoma, capazes de controlar a expressão gênica, sem alterar a sequência de DNA. Esse controle pode ocorrer por meio de modificações pós-traducionais de proteínas histonas, metilação de DNA ou expressão de microRNA, processos que causam instabilidade nas células e podem alterar a expressão gênica, provocando, eventualmente, a carcinogênese.51 A metilação do DNA representa a transferência de um grupo metil para o carbono 5 de um nucleotídeo, silenciando o gene que o contém, por meio da inibição da transcrição ou recrutamento de complexos correpressores da remodelação da cromatina. Eventos de metilação, principalmente em regiões promotoras de genes supressores de tumor ou relacionados ao tumor, já foram evidenciados na progressão de alguns tipos de câncer, como o melanoma, permitindo não só o desenvolvimento de biomarcadores epigenéticos com utilidade clínica, mas também a elucidação de sua tumorigênese.52 Alguns genes hipermetilados já foram descritos no CCM, como ilhas de CpG localizadas no promotor RASSF1A e o CDKN2A (p14ARF).13 O RASSF1A constitui um gene supressor de tumor que participa da regulação do ciclo celular e apoptose.53 Já a p14ARF é uma proteína codificada pelo gene CDKN2A, capaz de bloquear o ciclo celular nas fases G1 e G2 e de inibir o crescimento de células cancerígenas, ao ativar indiretamente o gene supressor de tumor p53.54 Dessa forma, o silenciamento desses genes observado no CCM sugere que as alterações na metilação podem estar diretamente envolvidas em sua carcinogênese.13 Além disso, apesar da infecção pelo MCPyV ser encontrada em cerca de 80% dos casos de CCM, possivelmente contribuindo para sua patogênese, há evidências de que a infecção viral não está relacionada com a inativação epigenética, o que indica a independência entre esses eventos.30,53 Em análises de amostras de pacientes diagnosticados com CCM, uma das linhagens celulares testadas, a MCC13, exibiu maior correspondência com o padrão de metilação do câncer de pulmão de pequenas células do que com o padrão encontrado no CCM. Como os perfis de metilação desses tipos de tumores são únicos, o resultado pode indicar que o CCM apresenta algumas células de origem derivadas de células metastáticas de outro tipo de carcinoma, que também possui características clínicas e patológicas semelhantes.13 A metilação do DNA também pode ser utilizada como um indicador da idade biológica de um indivíduo.55 Com isso, o desenvolvimento de relógios que estimam a idade epigenética, baseados na medida dos efeitos cumulativos das metilações ao longo dos anos, permite a melhor abordagem de questões como câncer e envelhecimento.56 Uma baixa idade de metilação do DNA é encontrada em células-tronco, e a indução de pluripotência está associada ao rejuvenescimento das células.57 Além disso, a aceleração da idade epigenética reflete a diferença entre a idade de metilação do DNA e a idade cronológica.55 Um estudo revelou que a idade de metilação do DNA de amostras de CCM era significativamente menor do que a idade cronológica dos pacientes analisados, independentemente da presença de MCPyV. A baixa idade de metilação do DNA pode indicar uma pluripotência dessas células, hipótese corroborada pela ideia da diferenciação trilinear do CCM, uma vez que o padrão de expressão concomitante de linhagens de células epiteliais, neuroendócrinas e de linfócitos pré e pró-B sugere as células-tronco como uma possível origem desse tumor. Entretanto, as amostras de CCM apresentaram resultado negativo ao serem submetidas à análise de pluripotência, revelando um comportamento paradoxal de juventude epigenética apesar da ausência de pluripotência.57
O CCM, descrito pela primeira vez em 1972, ainda não tem a sua origem definida com clareza, de modo que, até o momento, foram postuladas apenas hipóteses que explicam o seu desenvolvimento. A hipótese mais antiga tem como base as células derivadas da crista neural embrionária. Isso se deve a similaridades entre a carcinogênese do CCM e de tumores com origem definida a partir da crista neural, como a perda de genes supressores de tumor em loci gênicos semelhantes e a atividade telomerase evidenciada em ambos.11,44 Apesar da importância dessa semelhança, essa hipótese parece ser a menos provável atualmente, uma vez que outras similaridades não estão presentes nas células do CCM e nas células tumorais do melanoma e do neuroblastoma, por exemplo, tumores derivados da crista neural embrionária. Além disso, é possível que o CCM tenha origem a partir de células pré-B e pró-B, já que existem similaridades entre essas células e as células tumorais do câncer em questão. Tal semelhança decorre da presença de TdT, enzima característica de linfócitos em seu estágio de desenvolvimento inicial, em amostras do CCM.48 A presença de CD19 nas células do carcinoma também sustenta essa hipótese, uma vez que esse marcador é codificado a partir do gene PAX5, característico das células da linhagem linfocitária, também presente no tumor.49 Para os tumores em que é possível detectar o material genético do vírus, as células mais prováveis de se tornarem neoplásicas são os fibroblastos da derme localizados próximos aos folículos pilosos, uma vez que essas células são suscetíveis à infecção viral.2 Ao infectá-las, o poliomavírus integra seu material genético ao DNA celular, aumentando a taxa de proliferação celular, além de interferir em importantes vias de sinalização celular, o que pode acarretar o desenvolvimento do câncer.1,38 É possível, também, que o CCM tenha como origem células neoplásicas pré- existentes, que se desenvolvem a partir de células epiteliais. Essa hipótese tem como base a presença de mutações nos genes TP53 e Rb1, que são supressores tumorais, tanto no CCM quanto no carcinoma de células escamosas, na ausência de infecção pelo MCPyV.14 No caso de tumores vírus-positivo, a expressão do material genético viral concomitantemente à expressão de oncogenes pode favorecer a carcinogênese, teoria reforçada por casos já observados de amostras contendo características morfológicas e genéticas semelhantes entre o CCM e outros tumores cutâneos, como o tricoblastoma. Além disso, análises de linhagens celulares indicam que o padrão de metilação do CCM apresenta correspondências com outros tipos tumorais, como o câncer de pulmão de pequenas células, mostrando que o CCM pode ter origem em células metastáticas diversas, sem restringir-se às epiteliais. A epigenética é um importante fator para a elucidação do desenvolvimento do tumor. A hipótese que se baseia na modificação do genoma é sustentada pela observação, em amostras do CCM, de genes supressores de tumor silenciados a partir de mecanismos de hipermetilação.13 Ainda, análises epigenéticas tornaram improvável a ideia de células-tronco como uma possível origem do CCM, já que amostras desse tumor apresentaram resultado negativo quanto à pluripotência. Com base nos dados analisados, ainda não há informação suficiente disponível para se determinar a exata origem do CCM. Há uma ampla chance de que haja mais de uma origem celular para essa neoplasia, com a possível influência de mecanismos de modificação epigenética. Acreditamos que as células com maior probabilidade de originarem o CCM são células pouco diferenciadas, as quais podem se especificar em diferentes outros tipos celulares, o que é capaz de explicar a possível origem múltipla desse carcinoma. Apesar disso, alguns estudos, como o realizado por Chteinberg et al., apresentam resultados negativos para análise de pluripotência como a origem do CCM, fato que impede a definição objetiva da origem do CCM sem a realização de mais pesquisas neste âmbito.57
Fernanda Guimarães Souza
ORCID: 0009-0004-8385-0154
Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito
Daniela Pessanha dos Santos
ORCID: 0009-0002-1490-9613
Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Luiza Oliveira Ribeiro
ORCID: 0009-0009-7355-9378
Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Thiago Rubim Batista Bellot Nascimento
ORCID: 0000-0003-3909-5935
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Flávio Barbosa Luz
ORCID: 0000-0001-5454-8950
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
1. Uchi H. Merkell cell carcinoma: an update and immunotherapy. Front Ocol. 2018;8:48.
2. Krump NA, You J. From Merkel cell polyomavirus infection to Merkel cell carcinoma oncogenesis. Front Microbiol. 2021;12:739695.
3. Yang JF, You J. Merkel cell polyomavirus and associated Merkel cell carcinoma. Tumour Virus Res. 2022;13:200232.
4. Pietropaolo V, Prezioso C, Moens U. Merkel cell polyomavirus and Merkel cell carcinoma. Cancers (Basel). 2020;12(7):1774.
5. Toker C. Trabular carcinoma of the skin. Arch Dermatol.1972;105(1):107-110.
6. Bataille A, Le Gall C, Misery L, Talagas M. Merkel cells are multimodal sensory cells: a review of study methods. Cells. 2022;11(23):3827.
7. Tang CK, Toker C. Trabecular carcinoma of the skin: an ultrastructural study. Cancer. 1978;42(5):2311-2321.
8. Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. I. A clinopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9(2): 95-108.
9. Sunshine JC, Jahchan NS, Sage J, Choi J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene. 2018;37(11):1409-1416.
10. Tilling T, Moll I. Which are the cells of origin in Merkel cell carcinoma? J Skin Cancer. 2012;2012:680410.
11. Suárez C, Rodrigo JP, Ferlito A, Devaney KO, Rinaldo A. Merkel cell carcinoma of the head and neck. Oral Oncology. 2004; 40(8):773-779.
12. Kolhe R, Reid MD, Lee JR, Cohen C, Ramalingam P. Immunohistochemical expression of PAX5 and TdT by Merkel cell carcinoma and pulmonary small cell carcinoma: a potential diagnostic pitfall but useful discriminatory marker. Int J Clin Exp Pathol. 2013. 6(2):142-147.
13. Gujar H, Mehta A, Li HT, Tsai YC, Qiu X, Weisenberger DJ, et al. Characterizing DNA methylation signatures and their potential functional roles in Merkel cell carcinoma. Genome Med. 2021; 13(1):130.
14. Thibault K. Evidence of an epithelial origin of Merkel cell carcinoma. Mod Pathol. 2022;35(4):446-448.
15. Swann MH, Yoon J. Merkel cell carcinoma. Semin Oncol. 2007;34(1):51-56.
16. Hitchcock CL, Bland KI, Laney III RG, Franzini D, Harris B, Copeland EM. Neuroendocrine (Merkel cell) carcinoma of the skin. Its natural history, diagnosis, and treatment. Ann Surg. 1988;207(2):201-207.
17. Pectasides D, Pectasides M, Economopoulos T. Merkel cell cancer of the skin. Ann Oncol. 2006; 17(10):1489-1495.
18. Walsh NM, Cerroni L. Merkel cell carcinoma: a review. J Cutan Pathol. 2021;48(3):411-421.
19. Cerroni L, Kerl H. Primary cutaneous neuroendocrine ( Merkel cell) carcinoma in association with squamous – and basal – cell carcinoma. Am J Dermatopathol. 1997;19(6):610-613.
20. 20. Cornejo C, Miller CJ. Merkel cell carcinoma: updates on staging and management. Dermatol Clin. 2019;37(3):269-277.
21. Harms KL, Healy MA, Nghiem P, Sober AJ, Johnson TM, Bichakjian CK, et al. Analysis of prognostic factors from 9387 Merkel cell carcinoma cases forms the basis for the new 8th edition AJCC staging system. Ann Surg Oncol. 2016;23(11):3564-3571.
22. Baba PUF, Rasool Z, Younas Khan I, Cockerell CJ, Wang R, Kassir M, et al. Merkel cell carcinoma: from pathobiology to clinical management. Biology (Basel). 2021;10(12):1293.
23. Fried I, Cerroni L. [Merkel cell carcinoma]. Patholege. 2014;35(5):467-475.
24. Moll I, Kuhn C, Moll R. Cytokeratin 20 is a general marker of cutaneous Merkel cells while certain neuronal proteins are absent. J Invest Dermatol. 1995;104(6):910-915.
25. Eispert AC, Fuchs F, Brandner JM, Houdek P, Wladykowski E, Moll I. Evidence for distinct populations of human Merkel cells. Histochem Cell Biol. 2009;132(1):83-93.
26. Kurokawa M, Nabeshima K, Akiyama Y, Maeda S, Nishida T, Nakayama F, et al. CD56: a useful marker for diagnosing Merkel cell carcinoma. J Dermatol Sci. 2003;31(3):219-224.
27. Llombart B, Monteagudo C, Lopez-Guerrero JA, Carda C, Jorda E, Sanmartin O, et al. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology. 2005;46(6):622-634.
28. Shah IA, Netto D, Schlageter MO, Muth C, Fox I, Manne RK. Neurofilament immunoreactivity in Merkel-cell tumors: a differentiating feature from small-cell carcinoma. Mod Pathol. 1993;6(1):3-9.
29. Moll I, Roessler M, Brandner JM, Eispert AC, Houdek P, Moll R. Human Merkel cells – aspects of cell biology, distribution and functions. Eur J Cell Biol. 2005;84(2-3):259-271.
30. Feng H, Shuda M, Chang Y, Moore PS. Clonal Integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096-1100.
31. Wong SQ, Waldeck K, Vergara IA, Schröder J, Madore J, Wilmott JS, et al. UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. 2015;75(24):5228-5234.
32. DeCaprio JA. Molecular pathogenesis of Merkel cell carcinoma. Annu Rev Pathol. 2021;16:69-91.
33. Ahmed MM, Cushman CH, DeCaprio JA. Merkel cell polyomavirus: oncogenesis in a stable genome. Viruses. 2021;14(1):58.
34. Jaeger T, Ring J, Andres C. Histological, immunohistological, and clinical features of Merkel cell carcinoma in correlation to Merkel cell Polyomavirus status. J Skin Cancer. 2012;2012:983421.
35. Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5(3):e1000363.
36. Gjoerup O, Chang Y. Update on human polyomaviruses and cancer. Adv Cancer Res. 2010;106:1-51.
37. Csoboz B, Rasheed K, Sveinbjørnsson B, Moens U. Merkel cell polyomavirus and non‐Merkel cell carcinomas: guilty or circumstantial evidence? APMIS. 2020;128(2):104-120.
38. Starrett GJ, Marcelus C, Cantalupo PG, Katz JP, Cheng J, Akagi K, et al. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. mBio. 2017;8(1):e02079-16.
39. Nascimento TRBB. Detecção de poliomavírus de células de Merkel em carcinomas de queratinócitos pela imuno-histoquímica [dissertação]. [Niterói]: Universidade Federal Fluminense; 2023. 95 p.
40. Liu W, Yang R, Payne AS, Schowalter RM, Spurgeon ME, Lambert PF, et al. Identifying the target cells and mechanisms of Merkel cell polyomavirus infection. Cell Host Microbe. 2016;19(6):775-787.
41. Harms PW, Harms KL, Moore PS, DeCaprio JA, Nghiem P, Wong MKK, et al. The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nat Rev Clin Oncol. 2018;15(12):763-776.
42. Schoenwolf GC, Larsen WJ, Bleyl SB, Brauer PR, Francis-West PH. Larsen's Human Embryology. Churchill Livingstone.; 2009.
43. Munde P, Khandekar S, Dive A, Sharma A. Pathophysiology of Merkel cell. J Oral Maxillofac Pathol. 2013;17(3):408-412.
44. Harnett PR, Kearsley JH, Hayward NK, Dracopoli NC, Kefford RF. Loss of allelic heterozygosity on distal chromosome 1p in Merkel cell carcinoma. A marker of neural crest origins? Cancer Genet Cytogenet. 1991;54(1):109-113.
45. Kervarrec T, Aljundi M, Appenzeller S, Samimi M, Maubec E, Cribier B, et al. Polyomavirus-positive Merkel cell carcinoma derived from a Trichoblastoma suggests an epithelial origin of this Merkel cell carcinoma. J Invest Dermatol. 2020;140(5):976-985.
46. Urbanczyk S, Stein M, Schuh W, Jäck HM, Mougiakakos D, Mielenz D. Regulation of energy metabolism during early B lymphocyte development. Int J Mol Sci. 2018;19(8):2192.
47. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570-1580.
48. Buresh CJ, Oliai BR, Miller RT. Reactivity with TdT in Merkel cell carcinoma: a potencial diagnostic pitfall. Am J Clin Pathol. 2008;129(6):894-898.
49. Patton DT, Plumb AW, Abraham N. The survival and differentiation of pro- B and pre-B cells in the bone marrow is dependent on IL-7Rα Tyr449. J Immunol. 2014;193(7):3446-3455.
50. zur Hausen A, Rennspiess D, Winnepenninckx V, Speel EJ, Kurz AK. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry. Cancer Res. 2013;73(16):4982-4987.
51. Rotondo JC, Mazziotta C, Lanzillotti C, Tognon M, Martini F. Epigenetic dysregulations in Merkel cell polyomavirus-driven Merkel cell carcinoma. Int J Mol Sci. 2021;22(21):11464.
52. Greenberg ES, Chong KK, Huynh KT, Tanaka R, Hoon DSB. Epigenetic biomarkers in skin cancer. Cancer Lett. 2014;342(20):170-177.
53. Helmbold P, Lahtz C, Enk A, Herrmann-Trost P, Marsch WCh, Kutzner H, et al. Frequent occurrence of RASSF1A promoter hypermethylation and Merkel cell polyomavirus in Merkel cell carcinoma. Mol Carcinog. 2009;48(10):903-909.
54. Sunwoo HH, Suresh MR. The immunoassay handbook: theory and applications of ligand binding, ELISA and related techniques [Internet]. 4th ed. Elsevier; 2013. Part 9, Chapter 12, Cancers Markers; p 833-856.
55. Noroozi R, Ghafouri-Fard S, Pisarek A, Rudnicka J, Spólnicka M, Branicki W, et al. DNA methylation-based age blocks: from age prediction to age reversion. Ageing Res Rev. 2021;68:101314.
56. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115.
57. Chteinberg E, Vogt J, Kolarova J, Bormann F, Van Den Oord J, Speel EJ, et al. The curious case of Merkel cell carcinoma: epigenetic youth and lack of pluripotency. Epigenetics. 2020;15(12):1319-1324.
 All content the journal, except where identified, is under a Creative Commons Attribution-NonCommercial 4.0 International license - ISSN-e 1984-8773
All content the journal, except where identified, is under a Creative Commons Attribution-NonCommercial 4.0 International license - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}