


Bruna Margatho Elias; Giovanna Curi Campos; Hudson Dutra Rezende; José Roberto Paes Almeida; Karla Calaça Kabbach Prigenzi; Sandra Lopes Mattos Dinato
Data de submissão: 27/07/2021
Decisão Final: 18/08/2021
Fonte de financiamento: Nenhuma
Conflito de interesses: Nenhum
Como citar este artigo: Elias BM, Campos GC, Rezende HD, Almeida JRP, Prigenzi KCK, Dinato SLM. Carcinoma de células de Merkel: múltiplas lesões cutâneas primárias no membro inferior. Surg Cosmet Dermatol. 2022;14:e20210082
Carcinoma de células de Merkel é um tumor neuroendócrino raro e agressivo de pele que usualmente apresenta-se como lesão única na região de cabeça ou pescoço. Relata-se um caso de topografia e apresentação atípicas, com presença de múltiplos e simultâneos tumores na perna esquerda de rápida evolução, associados à linfonodomegalia inguinal palpável, com diagnóstico confirmado por meio de histopatologia e imuno-histoquímica. Realizada exérese de linfonodo inguinal esquerdo e das lesões cutâneas com margem de segurança.
Keywords: Carcinoma de Célula de Merkel; Células de Merkel; Poliomavírus das Células de Merkel
O carcinoma de células de Merkel (CCM) é uma forma rara de câncer de pele não melanoma de origem neuroendócrina, mais comum em homens brancos idosos.1 Em geral, manifesta-se como uma pápula ou nódulo assintomático, de cor rósea ou vermelho-azulada, com rápido aumento de tamanho, em semanas a meses.2 Os locais mais acometidos são as áreas fotoexpostas da cabeça e do pescoço.3 A patogênese do CCM ainda é desconhecida, apesar da associação com várias anormalidades cromossômicas, sinalização de crescimento e vias apoptóticas, além do possível envolvimento do poliomavírus na carcinogênese tumoral. Ainda hoje, o CCM representa verdadeiro desafio diagnóstico e terapêutico na prática clínica.
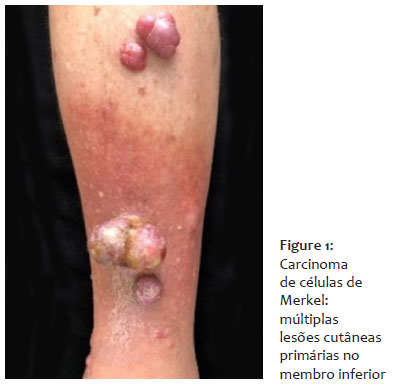
Paciente de 70 anos, caucasiano, sexo masculino, natural e procedente de Santos, aposentado, compareceu ao Setor de Dermatologia com queixa de múltiplas lesões de rápido crescimento na perna esquerda, há quatro meses. As lesões eram dolorosas, com exsudação incolor. O paciente referiu edema, mudança da coloração na região e dificuldade para deambular e negou quaisquer sintomas sistêmicos, apesar de fazer uso de atenolol e sinvastatina. Ao exame dermatológico, observaram-se múltiplas pápulas, nódulos, tumores (diâmetros variáveis de 1 a 6cm), com presença de exsudato. Na palpação, as lesões apresentavam consistência ligeiramente macia (Figura 1). Linfonodos inguinais bilaterais não aderidos e indolores eram palpáveis, e edema estava presente no membro comprometido. À dermatoscopia, visualizaram-se múltiplos vasos irregulares, polimorfos, áreas brancas brilhantes sem estruturas e áreas vermelho-leitosas. Além disso, foi notada lesão clinicamente compatível com carcinoma basocelular na região pré-esternal.
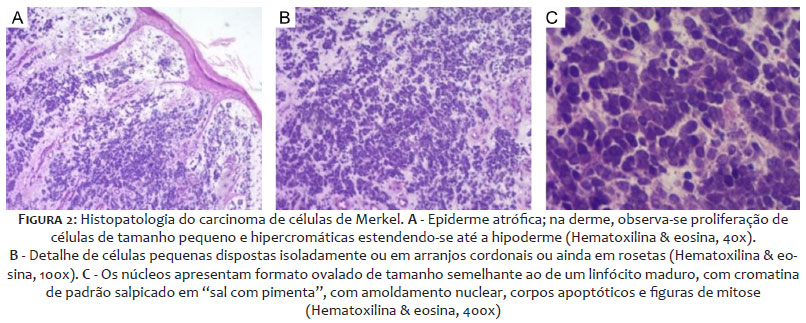
A biópsia de uma lesão papulosa solitária próxima ao maléolo medial esquerdo revelou proliferação de células pequenas atípicas na derme, com elevada relação núcleo/citoplasma, alto índice mitótico (>30 mitoses/10CGA) e numerosos corpos apoptóticos, dispostas em padrão cordonal ou blocos sólidos, que por vezes esboçavam rosetas, indicando uma neoplasia maligna de alto grau, sugerindo tratar-se de carcinoma neuroendócrino de pequenas células (Figura 2). A imuno-histoquímica foi solicitada e revelou imunoexpressão de citoqueratina 5/6, cromogranina, sinaptofisina e CK20 em padrão “dot”, confirmando a histogênese epitelial e a diferenciação neuroendócrina das células neoplásicas, compatíveis com o diagnóstico de CCM (carcinoma neuroendócrino de pequenas células primário da pele). Marcadores indicativos de diferenciação escamosa, de histogênese melanocítica e de sítio primário pulmonar e gastrointestinal também foram realizados e resultaram negativos.
Diante da raridade do caso com topografia e apresentação atípicas, foi realizada a pesquisa para imunossupressão, a qual não foi encontrada, sendo questionado o diagnóstico diferencial com carcinoma neuroendócrino de outro sítio primário. Avaliação complementar com tomografias computadorizadas (TCs) de tórax, abdômen e pelve não revelou doença em outros sítios, tampouco linfonodomegalia. A TC da perna esquerda mostrou lesões cutâneas exofíticas que mantinham continuidade com o tecido celular subcutâneo, sem apresentar plano nítido de clivagem com as fáscias musculares subjacentes e sem infiltrar músculos ou planos mais profundos. A pesquisa plasmática por PCR qualitativo do poliomavírus foi negativa.
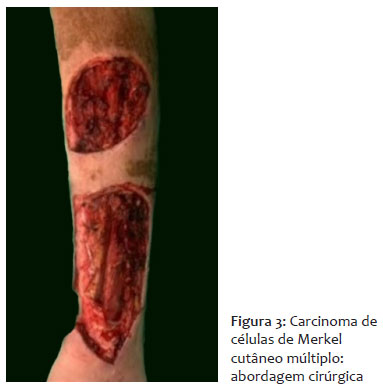
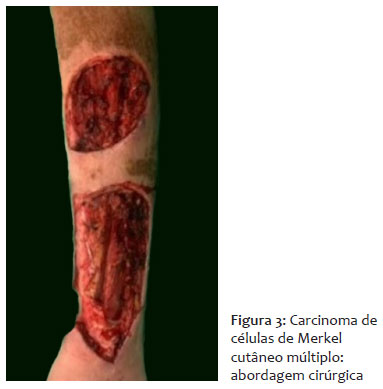
O paciente foi encaminhado para a cirurgia oncológica e submetido à exérese de linfonodo inguinal esquerdo, o qual foi enviado para congelação intraoperatória e histopatológico subsequente, com resultado negativo. Em seguida, foi realizada exérese das lesões cutâneas com margem de segurança (Figura 3).
Devido à extensa área exposta, com impossibilidade de fechamento primário e dificuldade para retalhos pela condição da pele, foi feito curativo com sulfadiazina de prata e atadura de Rayon, optando-se pela cicatrização por segunda intenção (Figura 4).
O CCM é raro e agressivo e afeta homens brancos na 7ª e 8ª décadas de vida, coincidindo com o perfil do paciente apresentado.1 Classicamente, envolve pele fotoexposta da região cervicofacial em 29-53% dos casos, segundo a literatura vigente. Aproximadamente 1/3 dos casos (35-38%) acomete as extremidades, sendo 21% membros superiores e somente 14-24% membros inferiores.4
O caso aqui relatado foi observado em região de raro acometimento. Em relação à apresentação clínica, esta é bastante variada, mas, tipicamente, é caracterizada por lesão nodular única eritêmato-violácea, persistente e assintomática, frequentemente menor que 2cm, de crescimento rápido, podendo chegar a 20cm em meses.1,5,6 Em quase 1/3 dos casos encontram-se outras neoplasias cutâneas simultâneas, como o carcinoma basocelular, fato concordante com o nosso paciente.3 Já a patogênese é incerta, mas o poliomavírus parece ter algum papel, já que é detectado na maioria dos casos (80%) e com frequência entre pacientes com síndrome da imunodeficiência adquirida e imunossuprimidos transplantados, estando integrado no genoma antes da expansão clonal das células tumorais, sugerindo ser um fator que contribui para o desenvolvimento do tumor.4,5 O paciente em questão não apresentava imunossupressão nem sorologia positiva para o vírus.
Devido ao quadro clínico usualmente inespecífico, a investigação com imagens é necessária, ajudando a diagnosticar e diferenciar o CCM de metástases de outros carcinomas neuroendócrinos, como o câncer de pulmão de pequenas células. O diagnóstico baseia-se em um exame minucioso da pele e dos linfonodos, biópsia e avaliação histológica por um dermatopatologista experiente.6 Histologicamente, apresenta-se como pequenas células basofílicas uniformes que preenchem a derme, com escasso citoplasma, cromatina “empoeirada” e moldagem nuclear. As células organizam-se em ninhos, espalhando-se para a derme reticular e subcutânea, com eventual envolvimento epidérmico. O CMM possui características neuroendócrinas e epiteliais, expressando alguns marcadores, sendo o CK204 um marcador bastante específico e sensível quando encontrado no padrão “dot” paranuclear, o que pode ser confirmado no caso apresentado.
Trata-se de um tumor altamente agressivo e metastático, com uma taxa de sobrevida específica da doença de 64% em cinco anos e de mortalidade de 33% em três anos após o diagnóstico.1,4 Sabe-se que 2/3 dos pacientes com CCM apresentam apenas doença local, mas doença nodal ou metastática no momento do diagnóstico não é incomum, e a taxa de recorrência local gira em torno de 25-33%.5 A biópsia de linfonodo sentinela, realizada nesse caso, deve ser considerada para todos os pacientes, pois 1/3 com doença clinicamente localizada no momento da apresentação tem envolvimento linfonodal oculto, 6 e metástases hematogênicas e/ou linfáticas distantes podem ocorrer envolvendo principalmente o fígado, ossos, cérebro e pele.1
Quanto ao tratamento do CCM, não há consenso. Excisão cirúrgica é a terapia padrão para lesões primárias menores que 3cm e radioterapia adjuvante para lesões maiores que 2cm. Alguns estudos recomendam margens de 2cm para tumores maiores que 2cm e margens de 1cm para tumores menores que 2cm. No entanto, não existem estudos controlados comparando diferentes margens de excisão, considerando a raridade da doença.1,4 Há evidências também para o uso da radioterapia exclusiva para doença irressecável, mas não para quimioterapia, reservada para casos de doença metastática.1,4
Apesar de raro, a incidência de CCM está aumentando devido ao avanço da idade da população, à maior exposição solar e ao aumento do número de indivíduos imunocomprometidos.7 Por se tratar de um tumor agressivo, as apresentações atípicas devem ser lembradas, uma vez que o reconhecimento precoce possibilita melhora da qualidade de vida e das perspectivas prognósticas.4, 7, 8
Bruna Margatho Elias 0000-0003-2615-5775
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura.
Giovanna Curi Campos 0000-0002-8155-4499
Elaboração e redação do manuscrito; revisão crítica da literatura.
Hudson Dutra Rezende 0000-0002-7039-790X
Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; participação efetiva na orientação da pesquisa; revisão crítica da literatura; revisão crítica do manuscrito.
José Roberto Paes Almeida 0000-0002-3869-6715
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito.
Karla Calaça Kabbach Prigenzi 0000-0002-8264-8972
Elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados.
Sandra Lopes Mattos Dinato 0000-0002-4547-0474
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dado; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
1. Bassi A, Arunachalam M, Galeone M, Scarfì F, Maio V, Moretti S, et al. Multiple clustered nodules on the leg. Diagnosis: Merkel cell carcinoma. J Clin Oncol. 2014;32(17):e61-2.
2. Almeida MW, Lopes CC, Almeida Junior HL, Costa LE. Carcinoma de células de Merkel em extremidade inferior [Merkel cell carcinoma in lower end]. Rev Col Bras Cir. 2012;39(2):165-7.
3. Fernández-Regueiro R, Suárez-Sánchez FJ, Morís-de la-Tassa J. Merkel cell carcinoma. Report of a case with an atypical location and presentation. Rev Esp Cir Ortop Traumatol (Engl Ed). 2019;63(4):313-5.
4. Blumenthal L, VandenBoom T, Melian E, Peterson A, Hutchens KA. Multiple primary Merkel cell carcinomas presenting as pruritic, painful lower leg tumors. Case Rep Dermatol. 2015;7(3):316-21.
5. Chatzinasiou F, Papadavid E, Korkolopoulou P, Levidou G, Panayiotides I, Theodoropoulos K, et al. An unusual case of diffuse Merkel cell carcinoma successfully treated with low dose radiotherapy. Dermatol Ther. 2015;28(5):282-6.
6. Coggshall K, Tello TL, North JP, Yu SS. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol. 2018;78(3):433-42.
7. Duprat JP, Landman G, Salvajoli JV, Brechtbühl ER. A review of the epidemiology and treatment of Merkel cell carcinoma. Clinics. 2011;66(10):1817-23.
8. Strobel ES, Feyer P, Steingräber M, Schmitt-Gräff A, Kohl PK. An unusual case of Merkel cell carcinoma. J Cancer Res Clin Oncol. 2008;134(2):119-23.
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}