


Kássila da Rosa Nasser1; Luiz Carlos Cuce2; Rossana de Farias Vasconcelos3; Ana Carolina Macedo4; Juliana Garcia Kako Rodriguez5; Rodrigo Garcia Arruda6; João Guilherme Finizola7
Relato do caso de paciente do sexo masculino com raro tumor de células granulares em braço esquerdo, comprovado com histopatológico e imuno-histoquímica (proteína S100 e CD68 positivas).
Keywords: BRAÇO; NEOPLASIAS CUTÂNEAS; TUMOR DE CÉLULAS GRANULARES
O tumor de células granulares (TCG), originalmente conhecido como tumor de Abrikossof é raro e de origem desconhecida.1
Constituido por células com citoplasma granular, tem características benignas, sendo raros os casos de malignidade. Pouco mais frequente em mulheres e negros, que acomete principalmente da terceira à quinta década de vida.1-3
Caracteriza-se clinicamente como nódulo solitário, assintomático, em geral na cabeça e no pescoço (de 45% a 65%). Destes, 70% são localizados na cavidade bucal (língua e mucosa oral), embora existam relatos de sua presença em outros órgãos.4
O diagnóstico se faz mediante exames histopatológico e imuno-histoquímico. A histologia é típica, mostrando nódulo mal definido, não encapsulado, com células poligonais, citoplasma granular abundante, núcleo pequeno e redondo e nucléolos proeminentes.
Na imuno-histoquímica as células são fortemente S-100 positivas.
Apresenta-se um caso desse tumor de localização rara e não descrita na literatura.
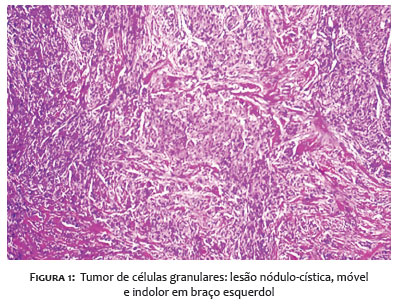
Paciente do sexo masculino com 30 anos apresentou-se à consulta com lesão nódulo-cística, de 1,3 x 0,8cm, assintomática no braço esquerdo há dois anos (Figura 1).
Foi realizada biópsia excisional da lesão, e o estudo histopatológico mostrou epiderme sem alterações e derme com proliferação neoplásica não encapsulada e circunscrita até a derme profunda, constituída por células em arranjo sólido, com amplo citoplasma eosinofílico granular e núcleos pequenos, regulares e monomórficos. Os anexos cutâneos apresentaram-se atróficos (Figura 2).
Na imuno-histoquímica as proteínas S100 (Figura 3) e a CD68 (Figura 4) apresentaram-se positivas difusamente - clone KP1. As demais proteínas, como p53, Melan-A, HMB45, desmina, CK5, CD34, AML, AE1+AE3, foram negativas, descaracterizando neoplasia melanocítica, epitelial, muscular ou vascular.
Há raros relatos de TCG na literatura, e as localizações típicas são cabeça e pescoço, sendo a mucosa oral a mais comum. Não foram encontrados na literatura relatos da presença desse tumor em membros superiores, como ocorreu no caso aqui descrito.
A origem ainda é desconhecida; no passado considerava-se essa célula proveniente de músculo liso, porém alguns estudos presumem sua origem na bainha neural, schwanniana, em que há positividade para a proteína S-100.5
Foram desenvolvidos nos últimos anos marcadores imuno-histoquímicos que são positivos para o TCG, com imunorreatividade positiva para alpha-1-antitripsina e CD68. O CD68 relaciona-se intimamente à glicoproteína da membrana lisossomal. A imunorreatividade positiva para alpha-1-antitripsina e CD68 no TCG pode ser reflexo do acúmulo intracitoplasmático dos fagolisossomas e não implica origem histiocítica para esses tumores.5,6 Do ponto de vista prático, portanto, apenas a coloração S-100 poderia ser usada para confirmação diagnóstica.
O tratamento do tumor é feito através de exerese cirúrgica, com margens adequadas, sendo o material enviado à patologia, como foi feito nesse caso. Nas raras ocorrências de multiplicidade desde tipo de tumor cutâneo, pode-se fazer a infiltração intralesional de glicocorticoides para reduzir o tamanho das lesões temporariamente, ocorrendo a regressão espontânea em alguns casos.7 Existe uma questão controversa em relação à malignidade desse tumor, desde que já existem casos relatados a respeito do TCG maligno. A variante maligna pode até mesmo emitir metástases, sendo mais comum em crianças e na região gengival.
Diante da localização atípica e ainda não publicada, e da raridade de casos relatados desse tumor, ressaltamos a necessidade de mais estudos para aprofundar conhecimentos sobre o assunto e a importância de exerese cirúrgica, uma vez que, eventualmente pode-se tratar de lesão maligna.
1. Brandão M, Domenech J, Noya M, Sampaio C, Almeida MVC, Guimarães NS, et al. Tumor de células granulares no pé (tumor de Abrikossoff): localização infreqüente de tumor relativamente raro. An Bras Dermatol. 2001;76(2):215-22.
2. Calonje E. Soft-Tissue Tumours and Tumour-like Conditions. In: Burns T, Breathnach S, Cox N, Griffiths C, eds. Rook's Textbook of Dermatology. 8th ed. Oxford: Wiley-Blackwell; 2010. p. 56.50.
3. Zangari F, Trombelli L, Calura G. Granular cell myoblastoma. Review of the literature and report of a case. Minerva Stomatol. 1996;45(5):231.
4. Ortiz-Hidalgo C, de la Vega G, Moreno-Collado C. Granular cell tumor (Abrikossoff tumor) of the clitoris. Int J Dermatol. 1997;36(12):926-37.
5. Filie AC, Lage JM, Azumi N. Immunoreactivity of S100 protein, alpha-1-antitrypsin, and CD68 in adult and congenital granular cell tumors. Mod Pathol. 1996;9(9):888-92.
6. Cavaliere A, Sidoni A, Ferri I, Falini B. Granular cell tumor: an immunohistochemical study. Tumori. 1994;80(3):224-8.
7. Baraf CS, Bender B. Multiple cutaneous granular cell myoblastoma. Arch Dermatol. 1964;89:243-6.
Trabalho realizado na Universidade de Santo Amaro - Santo Amaro (SP), Brasil.
 All content the journal, except where identified, is under a Creative Commons Attribution-NonCommercial 4.0 International license - ISSN-e 1984-8773
All content the journal, except where identified, is under a Creative Commons Attribution-NonCommercial 4.0 International license - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}