


Cíntia Ávila Souza1; Isabella Prado Motta1; Rodrigo Leite Azevedo1; Eliane Maria Ingrid Amstalden2; Thais Helena Buffo1
Fonte de financiamento: Nenhuma.
Conflito de interesses: Nenhum.
Data de Submissão: 20/09/2023
Decisão final: 22/10/2023
Como citar este artigo: Souza CA, Motta IP, Azevedo RL, Amstalden EMI, Buffo TH. Fibromixossarcoma mimetizando lesão benigna cística: relato de caso de tumor mesenquimal de alto grau em localização atípica. Surg Cosmet Dermatol. 2023;15:e20230305.
O fibromixossarcoma é um subtipo raro de sarcoma de partes moles, caracterizado pela presença de células fusiformes e matriz mixoide. Relatamos o caso de uma paciente idosa que apresentou crescimento progressivo de um nódulo interescapular, cuja investigação confirmou o diagnóstico de fibromixossarcoma de alto grau. É notório que esta neoplasia ocorre mais comumente nos membros inferiores e pode exibir recorrência associada ao aumento do grau histológico. Por isso, diagnóstico e tratamento precoces são fundamentais na mitigação da morbimortalidade. Enfatizamos a relevância do diagnóstico de um caso atípico e da intervenção terapêutica oportuna, contribuindo para a otimização dos desfechos clínicos.
Keywords: Sarcoma; Neoplasias de Tecido Conjuntivo e de Tecidos Moles; Tumores Fibrosos Solitários
O fibromixossarcoma é um subtipo raro de sarcoma de partes moles, composto por células fusiformes e estroma mixoide. É a neoplasia mesenquimal mais comum em pacientes idosos, apresentando-se como nódulos indolores, de crescimento lento, comum nas extremidades inferiores.1 Histologicamente, de acordo com a celularidade e atipia, podem ser classificados como de baixo, intermediário ou alto grau. Esta classificação não se correlaciona com o alto risco de recidiva (entre 50-60% para todos os subtipos), mas é um fator preditor de metástases, presentes de 20-35% nos graus intermediário e alto e incomum nos de baixo grau.2 Diferentemente dos demais sarcomas, o mixofibrossarcoma apresenta a tendência de, a cada recorrência, apresentar-se gradualmente com grau histológico mais elevado.3 Diante disso, percebe-se a importância do diagnóstico, estadiamento e tratamento precoces, visando a uma menor morbimortalidade. Relatamos o caso de uma paciente idosa com mixofibrossarcoma de alto grau na região interescapular.
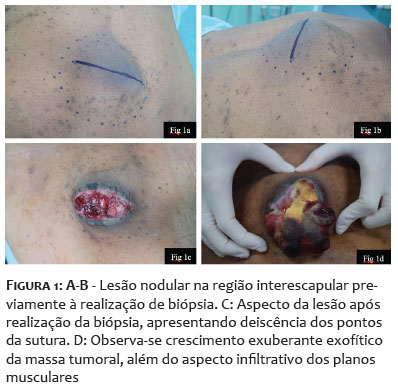
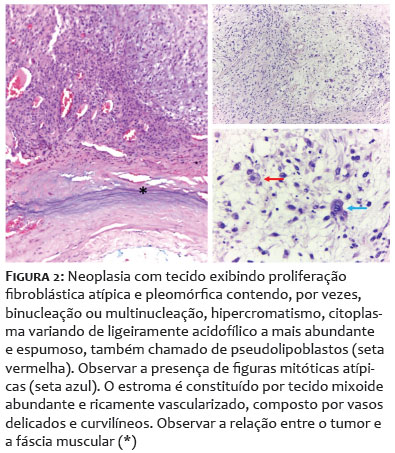
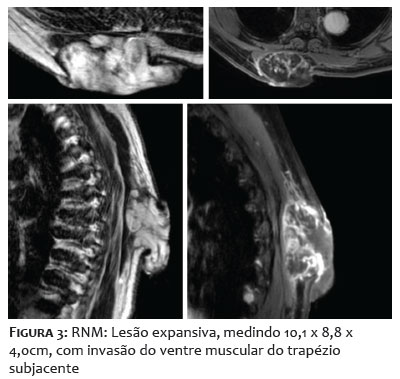
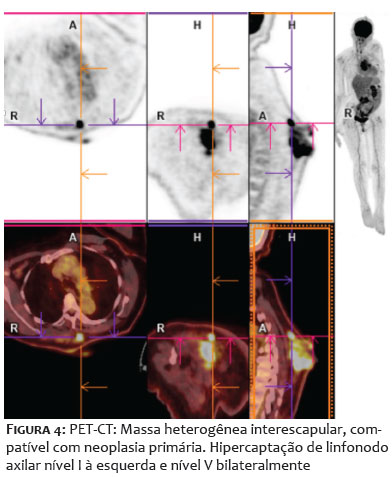
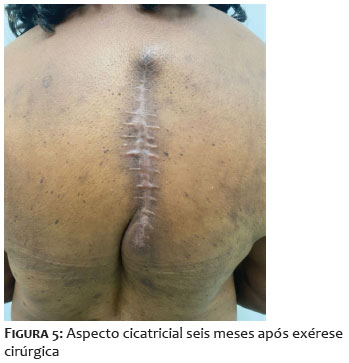
Paciente do sexo feminino, 68 anos, apresentou-se com lesão nodular indolor na região interescapular há um ano. Ao exame físico dermatológico inicial, observava-se lesão nodular, indolor, móvel, medindo 6 x 4cm. Apresentava USG de partes moles prévia, do início do quadro, compatível com cisto epidérmico com planos musculares livres (Figuras 1A-1B). Como a clínica não correspondia ao exame de imagem, foi solicitada tomografia, que demonstrou tratar-se de lesão cística, de 7 x 3cm de profundidade, sendo, então, agendada exérese. Após incisão, foi possível observar tecido amorfo, gelatinoso, enegrecido, não correspondendo à hipótese inicial aventada (Figuras 1C-1D). Realizada, então, biópsia profunda da lesão, com resultado histopatológico demonstrando mixofibrossarcoma de alto grau (Figura 2). Foi solicitada ressonância magnética de coluna, com presença de lesão expansiva, medindo 10,1 x 8,8 x 4,0cm, invadindo ventre muscular do trapézio subjacente, sem sinais de invasão dos demais planos musculares e das estruturas ósseas adjacentes (Figura 3). Para estadiamento, foi realizado PET-CT, que demonstrou hipermetabolismo em linfonodos axilares bilateral, sugestivos de metástases, o que não foi confirmado após biópsia dos linfonodos hipercaptantes (Figura 4). Paciente foi encaminhada à equipe da Ortopedia e foi realizada a exérese da lesão com margens amplas. Posteriormente, a paciente foi encaminhada à Oncologia para seguimento clínico, não sendo indicados tratamentos adjuvantes. Paciente mantém seguimento multidisciplinar, sem recidivas, após seis meses da exérese (Figura 5).
O fibromixossarcoma corresponde a um subtipo raro de sarcoma de partes moles, descrito pela primeira vez por Enzinger e Weiss em 1977.1 Previamente denominado histiocitoma fibroso maligno variante mixoide, apresenta origem mesenquimal, sendo composto por células fusiformes e estroma mixoide. É a neoplasia mesenquimal maligna mais comum em pacientes idosos, com predileção pela quinta a sétima décadas de vida, sendo o sexo masculino discretamente mais afetado.1-3
A localização preferencial é nos membros inferiores, sendo rara a ocorrência de lesões em tronco, membros superiores ou segmento cefálico. Em geral, apresentam-se como nódulos ou tumores indolores, de crescimento lento, normocrômicos ou discretamente eritematosos. Observa-se que sua ocorrência é maior em tecidos moles subcutâneos em relação aos tecidos profundos.1,4,5
O exame histopatológico é o padrão-ouro para estabelecer o diagnóstico. A amostra deve ser profunda para garantir uma avaliação fidedigna do material. Biópsias superficiais podem apresentar características benignas ou até mesmo subclassificar um tumor de alto grau.6 Histologicamente, é classificado como de baixo, intermediário ou alto grau, de acordo com a celularidade e atipia. A imuno-histoquímica apresenta baixa especificidade no mixofibrossarcoma sendo, em geral, positiva para vimentina e, em raros casos, para actina de músculo liso, S100 e desmina.6,7
A exérese da lesão com margens amplas é o tratamento de escolha. A recorrência local ocorre em 50-60% dos casos, e este risco parece não estar relacionado à profundidade das lesões ou ao grau histológico.8 Contudo, tem sido observado que, a cada recorrência, há uma tendência de o mixofibrossarcoma apresentar-se gradualmente mais celular, mais pleomórfico, mais mitoticamente ativo e, assim, com grau histológico mais elevado. Esta característica não é observada nos outros sarcomas. Em relação às metástases, elas são raras em tumores de baixo grau histológico, mas ocorrem entre 20-35% nos casos de tumores de intermediário e alto graus, notadamente para pulmões e ossos. O diagnóstico diferencial inclui outros tumores mixoides.1,2,8
A capacidade do tumor de apresentar altas taxas de recorrência e avançar em grau histológico sustenta a necessidade de um diagnóstico e tratamento precoces, objetivando uma menor morbimortalidade.
Cíntia Ávila Souza
ORCID: 0000-0001-8116-010X
Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Isabella Prado Motta 0009-0001-2858-578X
Concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura.
Rodrigo Leite Azevedo
ORCID: 0000-0001-8541-0591
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura.
Eliane Maria Ingrid Amstalden
ORCID: 0000-0001-7958-2426
Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados.
Thais Helena Buffo
ORCID: 0000-0002-6833-7596
Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; participação efetiva na orientação da pesquisa; revisão crítica do manuscrito.
1. Mentzel T, Calonje E, Wadden C, Camplejohn RS, Beham A, Smith MA, et al. Myxofibrosarcoma. Clinicopathologic analysis of 75 cases with emphasis on the low-grade variant. Am J Surg Pathol. 1996;20(4):391–405.
2. Mentzel T, Fletcher CDM. Low-Grade Myxofibrosarcoma. Pathology Case Reviews. 1998;3(3):139–42.
3. Mentzel T. Sarcomas of the skin in the elderly. Clin Dermatol. 2011;29(1):80–90.
4. Dore A, Robertson I, Williamson R, Weedon D. Progression of a myxoid pleomorphic fibroma to myxofibrosarcoma. Australas J Dermatol. 2003;44(4):287–90.
5. Nascimento AF, Bertoni F, Fletcher CDM. Epithelioid variant of Myxofibrosarcoma: expanding the clinicomorphologic spectrum of Myxofibrosarcoma in a series of 17 cases. Am J Surg Pathol. 2007;31(1):99–105.
6. Kosemehmetoglu K, Vrana JA, Folpe AL. Tumors of the soft tissues. In: Kumar V, Abbas AK, Aster JC, editors. Robbins and Cotran Pathologic Basis of Disease. 9th ed. Philadelphia: Elsevier; 2015. p. 1353-5.
7. Enzinger FM, Weiss SW. Soft tissue tumors. 3rd ed. St. Louis: CV Mosby, 1995.
8. Maretty-Kongstad K, Aggerholm-Pedersen N, Keller J, Safwat A. Recurrence and treatment outcome in 92 patients with fibromyxoid sarcoma from a single institution. Clin Orthop Relat Res. 2016;474(11):2523-2530.
 All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773
All content the journal, except where identified, under the Creative Commons Attribution 4.0 International licence - ISSN-e 1984-8773



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}